![]()
High Throughput Sequencing
- The high demand for low-cost sequencing has driven the development of high-throughput sequencing technologies that parallelize the sequencing process, producing thousands or millions of sequences at once.
- High-throughput sequencing is intended to lower the cost of DNA sequencing beyond what is possible with standard dye-terminator methods. In ultra-high-throughput sequencing, as many as 500,000 sequencing-by-synthesis operations may be run in parallel.
Pyrosequencing
- Pyrosequencing is a method of DNA sequencing (determining the order of nucleotides in DNA) based on the “sequencing by synthesis” principle, in which the sequencing is performed by detecting the nucleotide incorporated by a DNA polymerase.
- Pyrosequencing relies on light detection based on a chain reaction when pyrophosphate is released. Hence, the name pyrosequencing.
- The principle of Pyrosequencing was first described in 1993 by Bertil Pettersson, Mathias Uhlen and Pål Nyren by combining the solid phase sequencing method using streptavidin coated magnetic beads with recombinant DNA polymerase lacking 3´to 5´exonuclease activity (proof-reading) and luminescence detection using the firefly luciferase enzyme.
- A mixture of three enzymes (DNA polymerase, ATP sulfurylase and firefly luciferase) and a nucleotide (dNTP) are added to single stranded DNA to be sequenced and the incorporation of nucleotide is followed by measuring the light emitted.
- The intensity of the light determines if 0, 1 or more nucleotides have been incorporated, thus showing how many complementary nucleotides are present on the template strand.
- The nucleotide mixture is removed before the next nucleotide mixture is added.
- This process is repeated with each of the four nucleotides until the DNA sequence of the single stranded template is determined.
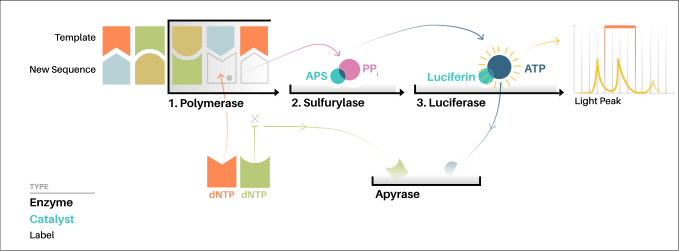
Procedure
- “Sequencing by synthesis” involves taking a single strand of the DNA to be sequenced and then synthesizing its complementary strand enzymatically.
- The pyrosequencing method is based on detecting the activity of DNA polymerase (a DNA synthesizing enzyme) with another chemoluminescent enzyme.
- Essentially, the method allows sequencing a single strand of DNA by synthesizing the complementary strand along it, one base pair at a time, and detecting which base was actually added at each step.
- The template DNA is immobile, and solutions of A, C, G, and T nucleotides are sequentially added and removed from the reaction. Light is produced only when the nucleotide solution complements the first unpaired base of the template.
- The sequence of solutions which produce chemiluminescent signals allows the determination of the sequence of the template.
- For the solution-based version of Pyrosequencing, the single-strand DNA (ssDNA) template is hybridized to a sequencing primer and incubated with the enzymes DNA polymerase, ATP sulfurylase, luciferase and apyrase, and with the substrates adenosine 5´ phosphosulfate (APS) and luciferin.
- The addition of one of the four deoxynucleotide triphosphates (dNTPs) (dATPαS, which is not a substrate for a luciferase, is added instead of dATP to avoid noise) initiates the second step.
- DNA polymerase incorporates the correct, complementary dNTPs onto the template. This incorporation releases pyrophosphate (PPi).
- ATP sulfurylase converts PPi to ATP in the presence of adenosine 5´ phosphosulfate. This ATP acts as a substrate for the luciferase-mediated conversion of luciferin to oxyluciferin that generates visible light in amounts that are proportional to the amount of ATP.
- The light produced in the luciferase-catalyzed reaction is detected by a camera and analyzed in a program.
- Unincorporated nucleotides and ATP are degraded by the apyrase, and the reaction can restart with another nucleotide.
The Illumina sequencing
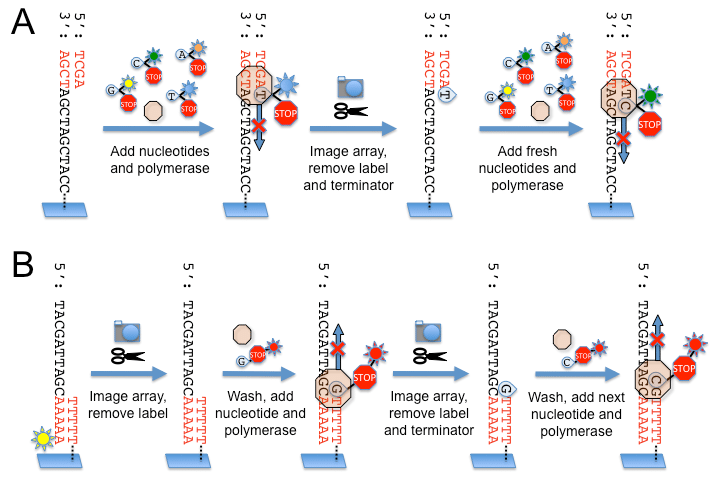
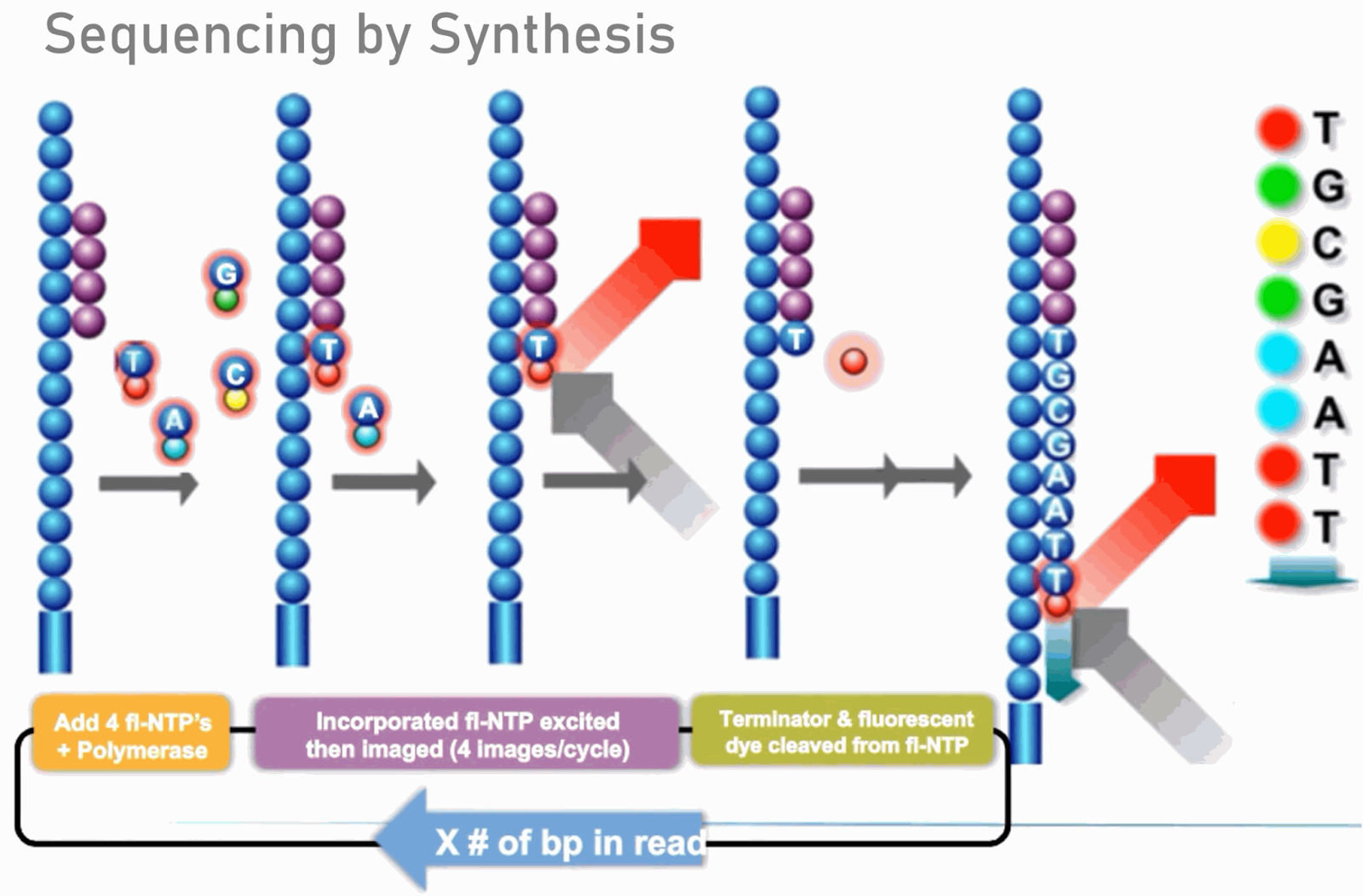
- The Illumina dye sequencing method is based on reversible dye-terminators and was developed in 1996 at the Geneva Biomedical Research Institute, by Pascal Mayer and Laurent Farinelli.
- In this method, DNA molecules and primers are first attached on a slide and amplified with polymerase so that local clonal colonies, initially coined “DNA colonies”, are formed.
- To determine the sequence, four types of reversible terminator bases (RT-bases) are added and non-incorporated nucleotides are washed away.
- Unlike pyrosequencing, the DNA chains are extended one nucleotide at a time and image acquisition can be performed at a delayed moment, allowing for very large arrays of DNA colonies to be captured by sequential images taken from a single camera.
- Decoupling the enzymatic reaction and the image capture allows for optimal throughput and theoretically unlimited sequencing capacity; with an optimal configuration, the ultimate throughput of the instrument depends only on the A/D conversion rate of the camera.
- The camera takes images of the fluorescently labeled nucleotides, then the dye along with the terminal 3′ blocker is chemically removed from the DNA, allowing the next cycle.
- In NGS, vast numbers of short reads are sequenced in a single stroke.
- To do this, firstly the input sample must be cleaved into short sections.
- The length of these sections will depend on the particular sequencing machinery used.
- In Illumina sequencing, 100-150bp reads are used. Somewhat longer fragments are ligated to generic adaptors and annealed to a slide using the adaptors.
- PCR is carried out to amplify each read, creating a spot with many copies of the same read. They are then separated into single strands to be sequenced.
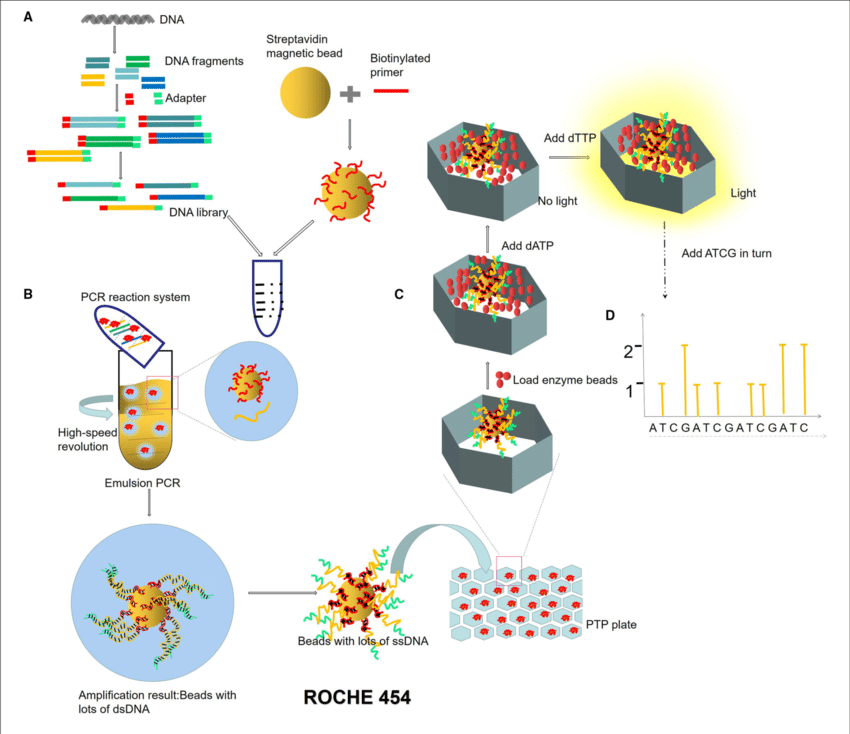
454 sequencing
- Roche 454 sequencing can sequence much longer reads than Illumina.
- Like Illumina, it does this by sequencing multiple reads at once by reading optical signals as bases are added.
- As in Illumina, the DNA or RNA is fragmented into shorter reads, in this case up to 1kb. Generic adaptors are added to the ends and these are annealed to beads, one DNA fragment per bead.
- The fragments are then amplified by PCR using adaptor-specifc primers.
- Each bead is then placed in a single well of a slide.
- So each well will contain a single bead, covered in many PCR copies of a single sequence.
- The wells also contain DNA polymerase and sequencing buffers.
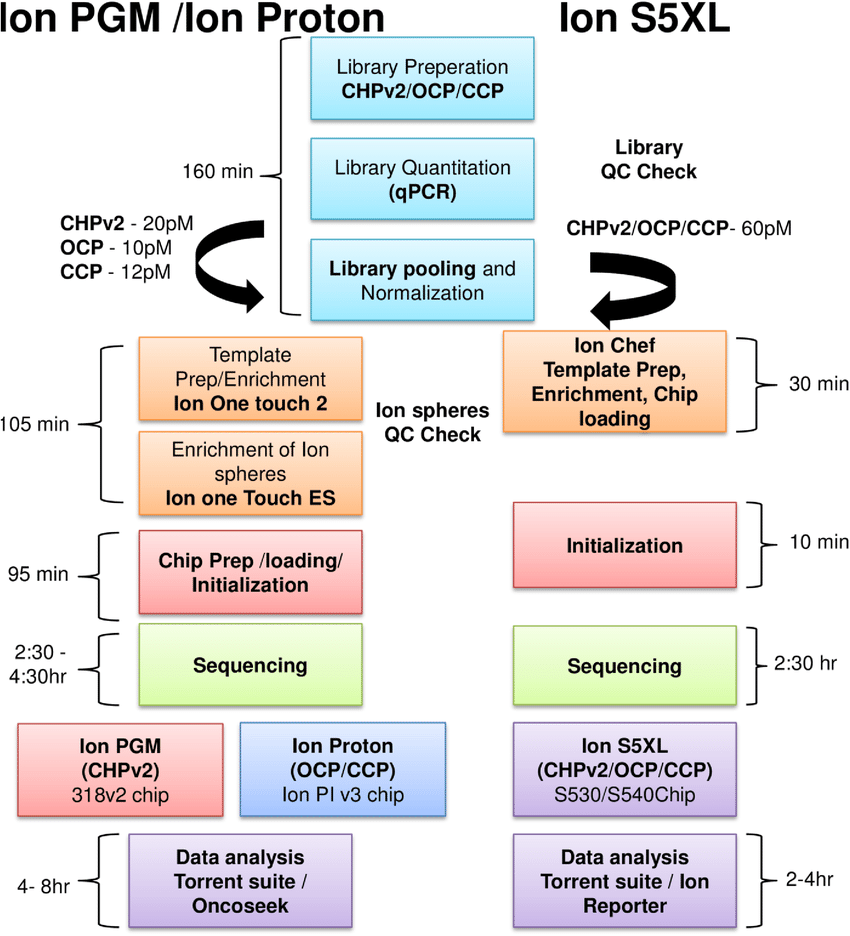
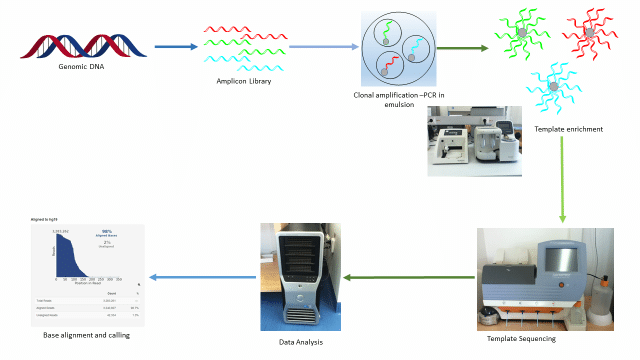
Ion Torrent: Proton / PGM sequencing
- Unlike Illumina and 454, Ion torrent and Ion proton sequencing do not make use of optical signals. Instead, they exploit the fact that addition of a dNTP to a DNA polymer releases an H+ ion.
- As in other kinds of NGS, the input DNA or RNA is fragmented, this time ~200bp. Adaptors are added and one molecule is placed onto a bead.
- The molecules are amplified on the bead by emulsion PCR. Each bead is placed into a single well of a slide.
Genome sequencing and variation
- The utility of HTS technologies for determining genome sequences de novo was first demonstrated by sequencing the genome of Acinetobacter baumannii.
- As the technologies and throughput improved, they were applied to “resequencing” human genomes and exomes, which was accomplished by first mapping reads to a reference genome and then identifying variants that differ between the sample genome and the reference .
- The different genome sequencing projects have since revealed that individuals typically harbor 3.5–4 million single nucleotide variants (SNVs) in total and several hundred thousand short indels relative to the reference genome. Importantly, these variants include hundreds of loss of function alterations in genes
- HTS has also been used to globally characterize structural variation (SV) in the human genome. SVs include large (>1kb) segments of the genome that have been duplicated, deleted or rearranged.
- The short read lengths of most HTS platforms make determining SVs and indels more challenging than SNVs .
- Typically, at least four independent approaches are utilized to identify SVs in a genome.
- These approaches include depth of read coverage, mapping of paired end reads that are discordant from the reference genome , identifying split reads and mapping of breakpoint junctions.
- Although each method has shortcomings, the improvement in resolution over array-based approaches has greatly enhanced our understanding of the prevalence of SVs throughout the genome and their contribution to disease.
- However, because no method or combination of them is comprehensive, SVs are never characterized in their entirety, if at all, in most sequencing projects.
- In addition to identifying variants, it is also useful to assign them to paternal and maternal alleles, or “phase” them. Similar to SVs, current read lengths hinder our ability to phase genomes.
- This limitation can be circumvented by several methods, including sequencing parents, sequencing proximity ligated fragments or dilution and barcoding strategies during template preparation to allow long read assembly.
- With approximately 30 Gbp of additional sequence data, ~99% of the SNVs identified in a 50x genome can be phased into blocks that are 0.2–1Mb in length. Understanding the phase of variants can have important clinical implications when determining if multiple damaging variants affect both copies of a gene or only one copy.
- To date, HTS has been applied to many thousands of genomes and many tens of thousands of exomes, yielding tremendous insight into human diversity and disease.
Mapping regulatory information of the genome
- HTS has applications beyond simply sequencing genomes. Perhaps one of the highest impact areas is the genome-wide mapping of DNA regulatory elements at high-resolution.
- The first of these technologies was ChIP-Seq in which DNA associated with a transcription factor (TF) or chromatin modification is immuno-selected and then sequenced using HTS.
- Mapping the sequences back to the genome reveals the location of bound regions or chromatin modifications.
- A more general method for discovering many putative regulatory regions is to map “open” regions of the genome using DNase I digestion, followed by DNA sequencing of the ends of the fragments .
- This method identifies approximately 50% of regions that are TF-bound as measured by ChIP-Seq.
- DNase-Seq, however, is quickly being replaced by Assay for Transposon Accessible Chromatin-Seq (ATAC-Seq) in which transposon-based insertion is used to map open chromatin regions with approximately 50 million mapped reads.
- The ATAC-seq protocol is also simpler and can be applied to small numbers of cells, even single cells.
- Regulatory information is especially revealing when compared across many individual genomes or within a single genome across many cell or tissue types.
- Large-scale application of these methods by the ENCODE project has provided a wealth of invaluable information regarding transcription factor binding networks , epigenetic maps and transcript annotations.
- Moreover, recent studies have found more than 3.5 million regulatory elements located throughout the genome in different cell types .
- One of the most striking findings from these studies as a whole, however, was the higher than expected portion of the genome that appears to be functional.
- The exact percentage is a source of significant debate , highlighting the importance of further experimental evidence to assign function to genomic elements.
- Genome targeting techniques, such as CRISPR-Cas9 as well as high-throughput enhancer assays provide researchers with new tools to interrogate putative regulatory elements.
- Nonetheless, a variety of lines of evidence (GWAS, ENCODE) suggest that the total amount of regulatory regions is likely greater than that of protein coding regions .
Mapping the three-dimensional organization of the genome
- Our understanding of the global organization and compartmentalization of chromosomes has been profoundly advanced by HTS technologies.
- 3D chromatin interactions can be studied using a variety of HTS assays, such as ChIA-PET (chromatin interaction analysis by paired-end tag sequencing) and Hi-C .
- Each of these assays relies upon proximity-based ligation of cross-linked, sheared chromatin followed by sequencing to derive contact maps.
- Hi-C was the first technique to allow unbiased, genome-wide interrogation of chromatin organization and revealed that the genome broadly partitions into open and closed chromatin states .
- Hi-C also demonstrated that the genome is organized into topological associating domains (TADs), which show high amounts of intra-domain interactions but exhibit infrequent interactions across domain boundaries .
- Interestingly, TAD organization is stable across cell types and evolutionarily conserved across species.
- The boundaries between TADs were also enriched for housekeeping genes and binding sites for the insulator protein CTCF, raising the possibility that the distribution of TADs is chromosomally encoded
- Recent advancements to the Hi-C technique combined with extremely deep sequencing (billions of reads per sample) have produced much higher resolution contact maps (~1kb), which refine TAD domain size from 1 Mb to less than 200 kbp .
- These new Hi-C maps demonstrated intrachromosomal looping events, often containing promoter-to-enhancer contacts that were associated with gene activation. Most loops were anchored with directionally-oriented CTCF binding sites, suggesting a mechanistic role for CTCF in establishing stable loops.
- Strikingly, fewer than 10,000 looping events were observed genome-wide, which is far smaller than previous estimates .
- Modeling of Hi-C data has also suggested a fractal globule chromatin state, a conformation that both maximizes packing while preserving the flexibility to access any genomic locus
Characterizing the transcriptome
- Our appreciation for the diverse cellular roles of RNA has been greatly enhanced by the advent of high-throughput sequencing.
- Much of this evolution in thought has been a direct result of the many HTS applications designed to systematically identify various classes of RNA as well as to characterize RNA structure, RNA-protein interactions and genomic localization.
- Cap analysis of gene expression (CAGE) and RNA-seq have been utilized to great effect to deeply characterize transcriptomes, providing precise, comprehensive measurements for message abundance, isoform usage, RNA-editing and allele-specific expression.
- Deep sequencing of RNA has suggested that roughly three-quarters of the human genome is transcribed .
- Most of this transcription covers introns or is very low, non-coding and of unclear biologic significance.
- However, many interesting species of non-coding RNA, including lncRNAs (long, non-coding), snoRNAs (small, nucleolar) and microRNAs, have been systematically described with RNA-seq and derivative techniques. A subset of lncRNAs, for example, have been revealed by overlaying RNA-seq data with ChIP-seq profiles characteristic of expressed genes .
- Building upon earlier cDNA sequencing and tiling array experiments, these HTS approaches expanded the list to include more than a thousand mammalian lncRNAs.
- Analogous expansions have occurred for many aspects of RNA biology, such as the number of sites undergoing RNA editing .
- Understanding the structure and biology of these newly discovered transcripts has led to the development of additional HTS applications. For instance, microRNA-target discovery has been facilitated by sequencing signatures of miRNA-mediated mRNA decay, using parallel analysis of RNA ends (PARE) .
- Furthermore, RNA immunoprecipitation chip (RIP-chip) and subsequently RIP-seq were utilized to show that approximately 20% of the lncRNAs associate with polycomb repressor complex 2 (PRC2), a chromatin-modifying complex Given these links to chromatin, methods analogous to ChIP-seq were developed, such as chromatin isolation by RNA purification (ChIRP-seq), to determine the genomic localization of lncRNAs .
- HTS applications have also made it possible to determine transcript structure both in vitro (parallel analysis of RNA sequencing; PARS) and in vivo (Structure-seq), providing insight into the effects of various structural features on translation efficiency, splicing and polyadenylation .
- More recently, systematic interrogation of sequence-function relationships for RNA-protein interactions has been made possible using a high-throughput biochemical assay called RNA on a massively parallel array (RNA-MaP) .
- The use of these assays, and many others, have enabled researchers to study RNA biology both comprehensively and with great detail, thereby enhancing our appreciation for the varied roles RNA plays in normal cellular homeostasis as well as human disease.
Microbiome sequencing
- Advances in HTS have enabled extensive cataloging of metagenomic samples, providing insight into the diversity of microbial species from a wide variety of sources, including the ocean, soil and the human body.
- These studies use both 16S rRNA gene sequencing to determine phylogenetic relationships as well as more comprehensive shotgun sequencing to predict detailed species and gene composition.
- In particular, much attention has been paid to characterizing the diverse microbes resident to healthy human populations .
- These studies found extensive variation in both body site habitat and among different individuals, giving rise to the concept of a “personal microbiome”.
- Microbial diversity, or the number and abundance distribution of microorganisms in a given niche, also correlates with several human diseases.
- For instance, an increase in diversity is associated with bacterial vaginosis, whereas obesity and inflammatory bowel disease exhibit a decrease in the diversity of gut microbes.
- Although transplant studies in mice have demonstrated a direct link between the gut microbiome, energy metabolism and obesity , causal relationships for the majority of human diseases are not well-established.
- A deeper understanding will require more detailed characterizations of the dynamics of microbiomes across health states as well as more integrative studies to investigate the functional interplay between the microbiota, the host and the environment.
Genome sequencing of rare diseases
- The capacity to sequence genomes, exomes and transcriptomes has profoundly influenced our understanding of the genetics of human disease, especially for rare Mendelian disorders and cancer.
- According to the Online Mendelian Inheritance in Man database, there are more than 7800 Mendelian disorders, but the causative gene for less than one half of these are known. By sequencing unrelated patients or affected and unaffected family members, early exome studies demonstrated the ability to identify causal alleles for a variety of inherited diseases .
- In rare cases, sequencing of patient samples has suggested specific clinical interventions that have dramatically altered patient outlook.
- In one early example, exome sequencing of a child with severe inflammatory bowel disease uncovered a mutation in an important regulator of inflammation, X-linked inhibitor of apoptosis (XIAP).
- Based on the severity of the child’s symptoms as well as the molecular diagnosis, a bone marrow transplant was given to the patient, which subsequently alleviate his symptoms.
- Despite the power of HTS for disease gene discovery, however, exome sequencing currently identifies the genetic defect in only 25% of cases
Cancer genome sequencing
- Cancer is another important arena where HTS has been applied to great effect. The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium (ICGC) have performed genome and exome sequencing on thousands of tumor-normal pairs.
- These studies have described the mutational landscapes for over 20 cancer types, demonstrating that tumors can vary dramatically in both the type and quantity of mutations.
- These global descriptions have been integral to the development of background mutation rates that are necessary for the detection of cancer driver genes.
- For example, replication timing and gene expression were both found to be important covariates when determining if a gene is mutated at a rate higher than expected .
- Using these background models, TCGA-led projects discovered several novel cancer drivers, known drivers in new cancer types and commonly disrupted pathways .
- Moreover, WGS of cancer samples has also identified high-frequency, non-coding mutations, such as activating mutations in the TERT promoter , a poorly characterized but highly relevant class of somatic variants.
- The scale and sensitivity of HTS has also enabled global descriptions of tumor heterogeneity, clonal evolution and the mechanisms underlying drug resistance.
- By tracking copy number aberrations in primary breast cancer cells using single-cell sequencing techniques,
- Navin and colleagues demonstrated that copy number rearrangements can occur in bursts, followed by persistent clonal expansion.
- Point mutations, in contrast, appear to accumulate more slowly over time, giving rise to more extensive clonal diversity, which may enable the tumor to adapt to diverse selective pressures .
- In addition to examining clonal diversity, HTS has also been used to compare primary tumors with relapse lesions, allowing characterization of the effects of chemotherapy as well as the molecular mechanisms underlying resistance to therapy .
- Together, these molecular portraits of cancer are forming the foundation of new paradigms for the diagnosis and treatment of cancer.
Limitations of current HTS technologies
- It is becoming increasingly clear that while the technologies of today may be capable of providing population-level sequencing to both researchers and clinicians, key limitations remain. From a technological perspective, accuracy and coverage across the genome are still problematic, particularly for GC-rich regions and long homopolymer stretches .
- In addition, the short read lengths produced by most current platforms severely limit our ability to accurately characterize large repeat regions, many indels and structural variation, leaving significant portions of the genome opaque or inaccurate .
- The establishment of a gold standard genome, as envisioned by the Genome in a Bottle Consortium as well as standards for data processing, variant calling and reporting as set out in the CLARITY Challenge , will be valuable for comparing and reporting the accuracy of different platforms and studies.
- Given the limitations and biases of different platforms, it is also likely that accurate genome sequencing will use a combination of technologies.
- In addition to genomes, quantitative analysis of complete transcriptomes, with individual allelic and spliced isoforms, is hindered by short reads as well as the cost and throughput of current long-read technologies. Improvements to current long read technologies, such as Pacific Biosciences and Oxford Nanopore Technologies, as well as the use of “synthetic long read methods” in which longer fragments can be sequenced and assembled from short reads will help overcome these limitations .
- Although both the research and medical communities are pressing forward with current technologies, these limitations will also continue to drive the innovation of new sequencing platforms (reviewed by
HTS in the coming era of personalized medicine
- To date, clinical HTS has most often been employed on focused regions of the genome or in the context of small pathogen identification.
- For instance, prenatal tests designed to non-invasively detect chromosomal abnormalities in cell-free DNA from maternal blood are clinically available (e.g., Ariosa Diagnostics’ Harmony Test and BGI’s NIFTY Test). Similarly, targeted HTS of clinically actionable mutations is being utilized to guide the diagnosis and treatment of cancer (e.g., Foundation Medicine’s FoundationONE test).
- HTS has also been employed in clinical contexts to monitor pathogen outbreaks, such as methicillin-resistant S. aureus infections .
- The development and use of these focused assays will continue to expand, but the full promise of personalized medicine relies upon the routine clinical application of more comprehensive techniques, such as WGS, which still faces significant challenges.
- In order for large-scale genomics to become fully integrated into the clinic, we need to reduce the costs and timescales associated with storage and interpretation of genome data.
- Most importantly, however, we must improve our ability to understand the biological and clinical consequences of variants of unknown significance.
- This class of alterations is the most common in personal genome sequences and includes novel variants that affect the coding sequence of known disease-causing genes but can also refer to variants in genes previously unlinked to disease or in regulatory regions of the genome. Interpretation of these variants will benefit from additional genome sequencing as well as the data provided by large-scale genomics projects, such as ENCODE and GTEx, which enable the generation of more complete reference databases.
- Open access projects, such as the Personal Genomes Project and integrative Personal Omics Profiling (iPOP) will also provide valuable community resources for linking phenotypes to sequences .
- The incorporation of high-throughput biochemical measurements of novel variation and detailed health records along with open data sharing will maximize our ability to both interpret personal genomes and better understand human health and disease.

Related posts:
 Artificial chromosomes
Artificial chromosomes
 Recombinant DNA Technology Tools and Technique of rDNA Tech
Recombinant DNA Technology Tools and Technique of rDNA Tech
 Genetic Engineering of Plants: Aims strategies for development of transgenic
Genetic Engineering of Plants: Aims strategies for development of transgenic
 Molecular Marker
Molecular Marker
 Proteomics
Proteomics
 Concept of Cellular differentiation totipotency
Concept of Cellular differentiation totipotency
 Protoplast Regeneration
Protoplast Regeneration
 Genetic and physical Mapping of Gene
Genetic and physical Mapping of Gene


