![]()
Recombinant DNA Technology Tools and Technique of rDNA Tech
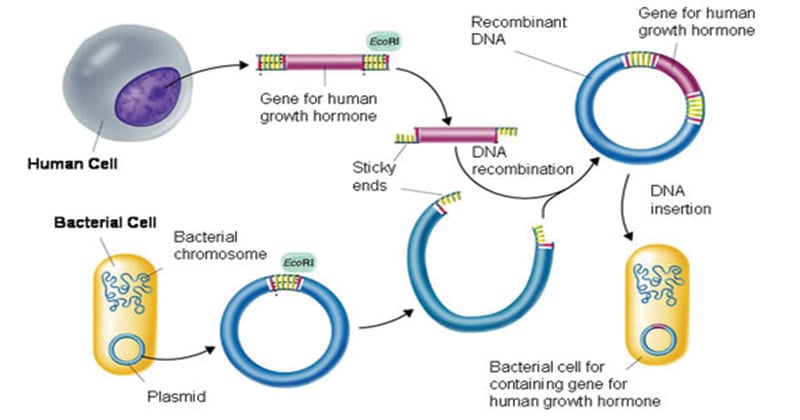
- Recombinant DNA technology refers to the joining together of DNA molecules from two different species that are inserted into a host organism to produce new genetic combinations that are of value to science, medicine, agriculture, and industry.
- Recombinant DNA (rDNA), on the other hand is the general name for a piece of DNA that has been created by the combination of at least two strands.
- They are DNA molecules formed by laboratory methods of genetic recombination (such as molecular cloning) to bring together genetic material from multiple sources, creating sequences that would not otherwise be found in the genome.
- Recombinant DNA in a living organism was first achieved in 1973 by Herbert Boyer, of the University of California at San Francisco, and Stanley Cohen, at Stanford University, who used E. coli restriction enzymes to insert foreign DNA into plasmids. Steps of Genetic Recombination Technology
Basic steps involved in rDNA technology (or genetic engineering) are given below
- Selection and isolation of DNA insert
- Selection of suitable cloning vector
- Introduction of DNA-insert into vector to form rec DNA molecule
- rec DNA molecule is introduced into a suitable host.
- Selection of transformed host cells.
- Expression and multiplication of DNA-insert in the host.
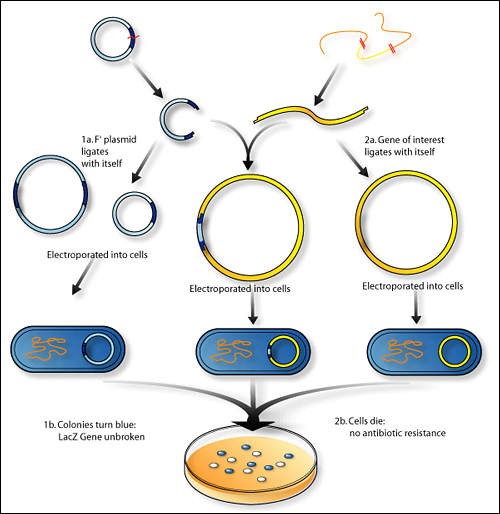
(i) Selection and isolation of DNA insert:
- First step in rec DNA technology is the selection of a DNA segment of interest which is to be cloned.
- This desired DNA segment is then isolated enzymatically.
- This DNA segment of interest is termed as DNA insert or foreign DNA or target DNA or cloned DNA.
(ii) Selection of suitable cloning vector:
- A cloning vector is a self-replicating DNA molecule, into which the DNA insert is to be integrated.
- A suitable cloning vector is selected in the next step of rec DNA technology.
- Most commonly used vectors are plasmids and bacteriophages.
(iii) Introduction of DNA-insert into vector to form recDNA molecule:
- The target DNA or the DNA insert which has been extracted and cleaved enzymatically by the selective restriction endonuclease enzymes
- [in step (i)] are now ligated (joined) by the enzyme ligase to vector DNA to form a rec DNA molecule which is often called as cloning-vector-insert DNA construct.
(iv) rec DNA molecule is introduced into a suitable host:
- Suitable host cells are selected and the rec DNA molecule so formed [in step (iii)] is introduced into these host cells.
- This process of entry of rec DNA into the host cell is called transformation.
- Usually selected hosts are bacterial cells like E. coli, however yeast, fungi may also be utilized.
(v) Selection of transformed host cells:
- Transformed cells (or recombinant cells) are those host cells which have taken up the recDNA molecule.
- In this step the transformed cells are separated from the non-transformed cells by using various methods making use of marker genes.
(vi) Expression and Multiplication of DNA insert in the host:
- Finally, it is to be ensured that the foreign DNA inserted into the vector DNA is expressing the desired character in the host cells.
- Also, the transformed host cells are multiplied to obtain sufficient number of copies.
- If needed, such genes may also be transferred and expressed into another organism.
Tools for Recombinant DNA Technology:
rDNA technology utilizes a number of biological tools to achieve its objectives, most important of them being the enzymes.
Important biological tools for rec DNA technology are:
(A) Enzymes:
a. Restriction Endonucleases
b. Exonucleases
c. DNA ligases
d. DNA polymerase
(B) Cloning Vector
(C) Host organism
(D) DNA insert or foreign DNA
(E) Linker and adaptor sequences.
(A) ENZYMES:
A number of specific enzymes are utilized to achieve the objectives of rec DNA technology.
The enzymology of genetic engineering includes the following types of enzymes:
(a) Restriction Endonuclease:
- The presence of restriction enzymes was first of all reported by W. Arber in 1962. He found that when the DNA of a phage was introduced into a host bacterium, it was fragmented into small pieces.
- This led him to postulate the presence of restriction enzymes. The first true restriction endonuclease was isolated in 1970s from the bacterium E. coli by Meselson and Yuan.
- Another important breakthrough was the discovery of restriction enzyme Hind-II in 1970s by Kelly, Smith and Nathans.
- They isolated it from -the bacterium Haemophilus influenza. In the year 1978, the Nobel Prize for Physiology and Medicine was given to Smith, Arber and Nathans for the discovery of endonucleases.
- These enzymes serve as important tools to cut DNA molecules at specific sites, which is the basic need for rec DNA technology.
- These are the enzymes that produce internal cuts (cleavage) in the strands of DNA, only within or near some specific sites called recognition sites/recognition sequences/ restriction sites or target sites. Such recognition sequences are specific for each restriction enzyme.
- Restriction endonuclease enzymes are the first necessity for rec DNA technology.
Types of Restriction Endonucleases:
There are 3 main categories of restriction endonuclease enzymes:
Type-I Restriction Endonucleases
Type-II Restriction Endonucleases
Type-III Restriction Endonucleases
Type-I Restriction Endonucleases:
- These are the complex types of endonucleases which cleave only one strand of DNA. These enzymes have the recognition sequences of about 15 bp length (Table 1).
- They require Mg++ ions and ATP for their functioning. Such types of restriction endonucleases cleave the DNA about 1000 bp away from the 5′ end of the sequence ‘TCA’ located within the recognition site.
- Important examples of Type-I restriction endonuclease enzymes are EcoK, EcoB, etc.
Type-II Restriction Endonucleases:
- These are most important endonucleases for gene cloning and hence for rec DNA technology. These enzymes are most stable.
- They show cleavage only at specific sites and therefore they produce the DNA fragments of a defined length.
- These enzymes show cleavage in both the strands of DNA, immediately cuts.the recognition sequences.
- They require Mg++ ions for their functioning.
- Such enzymes are advantageous because they don’t require ATP for cleavage and they cause cleavage in both strands of DNA.
- Only Type II Restriction Endonucleases are used for gene cloning due to their suitability.
- The recognition sequences for Type-II Restriction Endonuclease enzymes are in the form of palindromic sequences with rotational symmetry, i.e., the base sequence .n the first half of one strand of DNA is the mirror image of the second half of other strand of that DNA double helix .
- Important examples of Type-II Restriction endonucleases include Hindl, EcoRI, PvuII, Alul, Haelll etc.
Type-III Restriction Endonucleases:
- These are not used for gene cloning.
- They are the intermediate enzymes between Type-I and Type-II restriction endonuclease.
- They require Mg++ ions and ATP for cleavage and they cleave the DNA at well-defined sites in the immediate vicinity of recognition sequences, e.g. Hind III, etc.
Nature of cleavage by Restriction Endonucleases:
The nature of cleavage produced by a restriction endonuclease is of considerable importance.
They cut the DNA molecule in two ways:
1. Many restriction endonucleases cleave both strands of DNA simply at the same point within the recognition sequence. As a result of this type of cleavage, the DNA fragments with blunt ends are generated. PvuII, Haelll, Alul are the examples of restriction endonucleases producing blunt ends. Blunt ends may also be referred to as flush ends.
2. In the other style of cleavage by the restriction endonucleases, the two strands of DNA are cut at two different points. Such cuts are termed as staggered cuts and this results into the generation of protruding ends i.e., one strand of the double helix extends a few bases beyond the other strand. Such ends are called cohesive or sticky ends.
Such ends have the property to pair readily with each other when pairing conditions are provided. Another feature of the restriction endonucleases producing such sticky ends is that two or more of such enzymes with different recognition sequences may generate the same sticky ends.
(b) Exonucleases:
- Exonuclease is an enzyme that removes nucleotides from the ends of a nucleic acid molecule. An exonuclease removes nucleotides from the 5′ or 3′ end of a DNA molecule. An exonuclease never produces internal cuts in DNA.
- In rec DNA technology, various types of exonucleases are employed like Exonuclease Bal31, E. coli exonuclease III, Lambda exonuclease, etc.
- Exonuclease Bal31 are employed for making the DNA fragment with blunt ends shorter from both its ends.
- E coli Exonuclease III is utilized for 3’end modifications because it has the capability to remove nucleotides from the 3′-OH end of DNA.
- Lambda exonuclease is used to modify 5′ ends of DNA as it removes the nucleotides from 5′ terminus of a linear DNA molecule.
(c) DNA ligase:
- The function of these enzymes is to join two fragments of DNA by synthesizing the phosphodiester bond.
- They function to repair the single stranded nicks in DNA double helix and in rec DNA technology they are employed for sealing the nicks between adjacent nucleotides. This enzyme is also termed as molecular glue.
(d) DNA polymerases:
- These are the enzymes which synthesize a new complementary DNA strand of an existing DNA or RNA template.
- A few important types of DNA polymerases are used routinely in genetic engineering. One such enzyme is DNA polymerase ! which , prepared from E coli.
- The Klenow fragment of DNA polymerase-I .s employed to make the protruding ends double-stranded by extension of the shorter strand.
- Another type of DNA polymerase used in genetic engineering is Taq DNA polymerase which is used in PCR (Polymerase Chain Reaction).
- Reverse transcriptase is also an important type of DNA polymerase enzyme for genetic engineering. It uses RNA as a template for synthesizing a new DNA strand called cDNA a e complementary DNA).
- Its main use is in the formation of cDNA libraries.
- Apart from all these above mentioned enzymes, a few other enzymes also mark their importance in genetic engineering.
A brief description of these is given below:
(a) Terminal deoxynucleotidyl transferase enzyme:
- It adds single stranded sequences to 3′-terminus of the DNA molecule.
- One or more deoxyribonucleotides (dATP, dGTP, dl IP, dCTP) are added onto the 3′-end of the blunt-ended fragments.
(b) Alkaline Phosphatase Enzyme:
- It functions to remove the phosphate group from the 5′-end of a DNA molecule.
(c) Polynucleotide Kinase Enzyme:
- It has an effect reverse to that of Alkaline Phosphatase, i.e. it functions to add phosphate group to the 5′-terminus of a DNA molecule
(B) Cloning Vectors:
- It is another important natural tool which geneticists use in rec DNA technology.
- The cloning vector is the DNA molecule capable of replication in a host organism, into which the target DNA is introduced producing the rec DNA molecule.
- A cloning vector may also be termed as a cloning vehicle or earner DNA or simply as a vector or a vehicle a great variety of cloning vectors are present for use with E. coli is the host organism.
- However under certain circumstances it becomes desirable to use different hosts for cloning experiments. So, various cloning vectors have been developed based on other bacteria like Bacillus, Pseudomonas, Agrobacterium, etc. and on different eukaryotic organisms like yeast and other fungi.
- The cloning vector which has only a single site for cutting by a particular restriction endonuclease is Considered as a good cloning vector.
- Different types of DNA molecules may be used as cloning vehicles such as they may be plasmids, bacteriophages, cosmids, phasmids or artificial chromosomes.
(C) Host Organism:
- A good host organism is an essential tool tor genetic engineering.
- Most widely used host for rec DNA technology is the bacterium E. coli. because cloning and isolation of DNA inserts is very easy in this host.
- A good host organism is the one winch easy to transform and in which the replication of rec DNA is easier.
- There should not be any interfering element against the replication of rec DNA in the host cells
(D) DNA Insert Or Foreign DNA:
- The desired DNA segment which is to be cloned is called as DNA insert or foreign DNA or target DNA.
- The selection of a suitable target DNA is the very first step of rec DNA technology.
- The target DNA (gene) may be of viral, plant, animal or bacterial origin.
Following points must be kept in mind while selecting the foreign DNA:
1. It can be easily extracted from source.
2. It can be easily introduced into the vector.
3. The genes should be beneficial from a commercial or research point of view.
A number of foreign genes are being cloned for the benefit of human beings. Some of these DNA inserts are the genes responsible for the production of insulin, interferon, lymphotoxins, various growth factors, interleukins, etc.
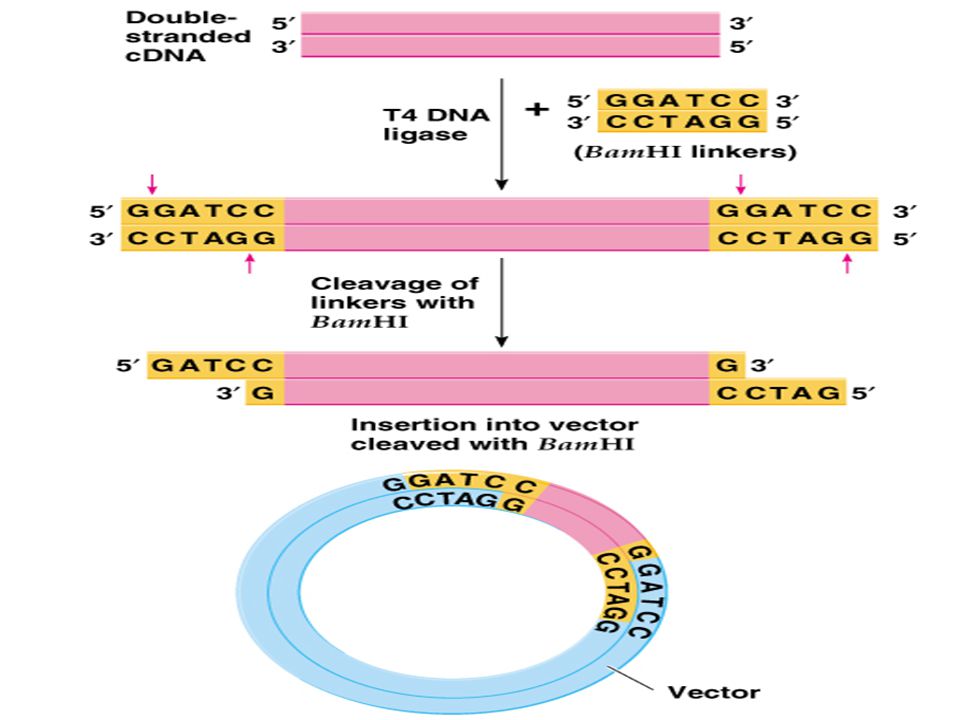
(E) Linker and Adaptor Sequences:
- Linkers and adaptors are the DNA molecules which help in the modifications of cut ends of DNA fragments. These can be joined to the cut ends and hence produce modifications as desired.
- Both are short, chemically synthesized, double stranded DNA sequences. Linkers have (within them) one or more restriction endonuclease sites and adaptors have one or both sticky ends. Different types of linkers and adaptors are used for different purposes.
- Linkers contain target sites for the action of one or more restriction enzymes. They can be ligated to the blunt ends of foreign DNA or vector DNA .
- Then they undergo a treatment with a specific restriction endonuclease to produce cohesive ends of DNA fragments EcoRI-linker is a common example of frequently used linkers.
- Adaptors are the chemically Synthesized molecules which have pre-formed cohesive ends . Adaptors are employed for end modification in cases where the recognition site for restriction endonuclease enzyme is present within the foreign DNA.
- The foreign DNA is ligated with an adaptor on both ends. This new molecule, so formed, is then phosphorylated at the 5′-termini. Finally foreign DNA modified with adaptors is integrated into the vector DNA to form the recombinant DNA molecule.
Techniques Used In Recombinant DNA Technology:
- A number of techniques are used for various purposes during different steps of rec DNA technology.
- Such techniques serve for the fulfilment of different requirements or to obtain proper information for drawing an exact inference during genetic engineering.
- Some of these important techniques are gel electrophoresis, blotting techniques, dot-blot hybridization, DNA sequencing, artificial gene synthesis, polymerase chain reaction, colony hybridization, etc.
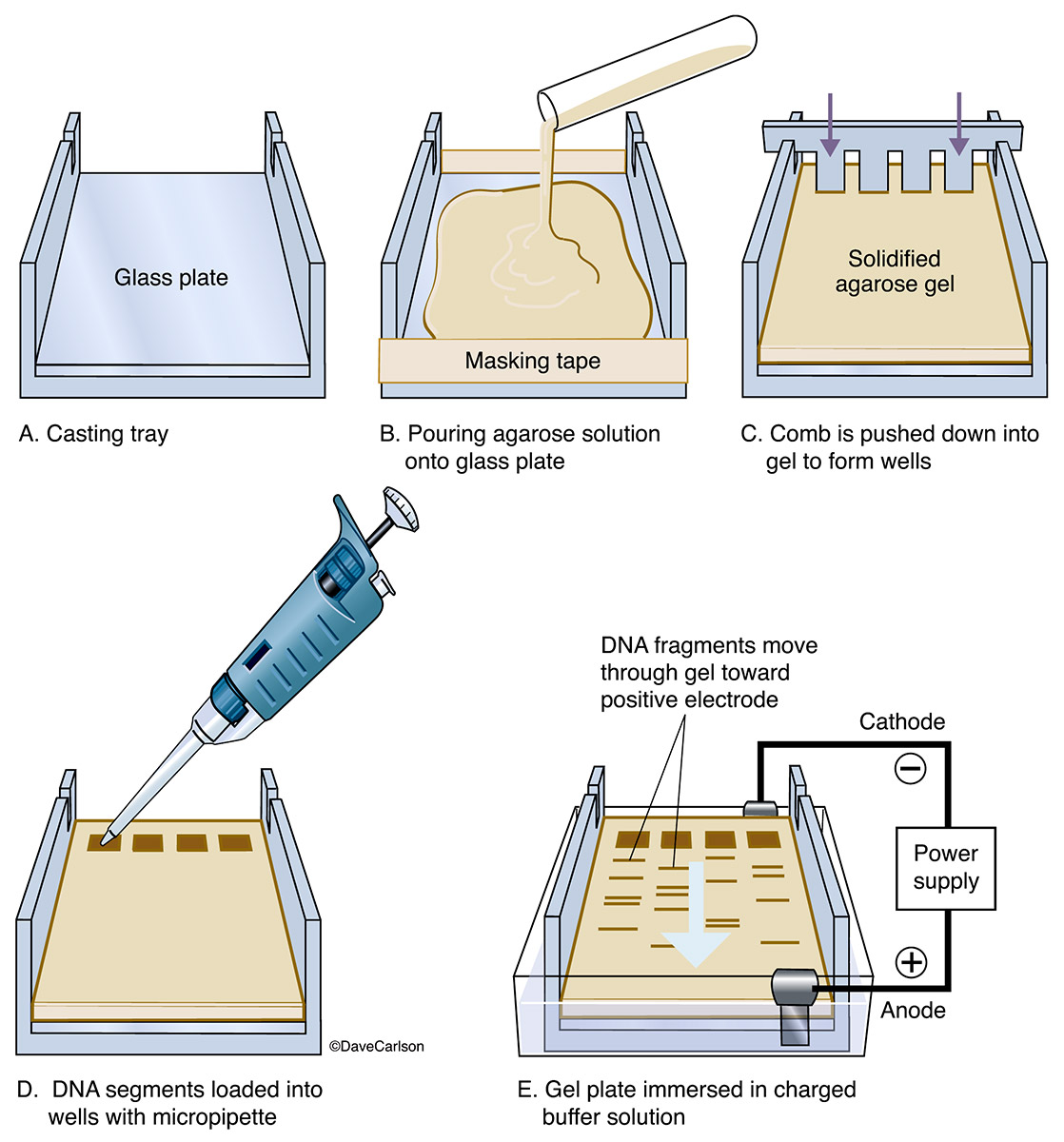
Gel Electrophoresis:
- It is the technique of separation of charged molecules (in aqueous phase) under the influence of an electrical field so that they move on the gel towards the electrode of opposite charge i.e., cations move towards the negative electrode and anions move towards the positive electrode.
- The genomic DNA is extracted from the desired host and is then fragmented using restriction endonucleases.
- For separation of these cut fragments and isolation of desired DNA fragments, the technique of gel electrophoresis is employed. Gel electrophoresis may be of horizontal or vertical type. Usually agarose gel is used for separation of large segments of DNA while the polyacrylamide gel is used for the separation of small DNA fragments which are only a few base pairs long.
- Gel electrophoresis employs a buffer system, a medium which is a gel and a source of direct current . Samples having DNA fragments are applied on the gel and current is passed through the system for an appropriate time. different DNA fragments move up to different distances on the gel depending on their charge to mass ratio.
- The heavier fragments move a little, while the lighter DNA fragments move up to a larger distance. Following the migration of the molecules, the gel is treated with selective stains to show the location of separated molecules in the form of bands.
- Very large DNA molecules or chromosomes cannot be separated even by Agarose Gel electrophoresis. For separation of such very large DNA molecules (sometimes representing whole chromosomes), a new technique is used which is known as Pulse Field Gel Electrophoresis (PFGE).
Blotting Techniques:
Visualization of a specific DNA (or RNA or protein) fragment out of many molecules requires a technique called blot transfer. In this technique, the separated bands are transferred onto a nitrocellulose membrane from the gel.
Mainly there are three types of blot transfer procedures:
Southern Blotting, Northern Blotting and Western blotting.
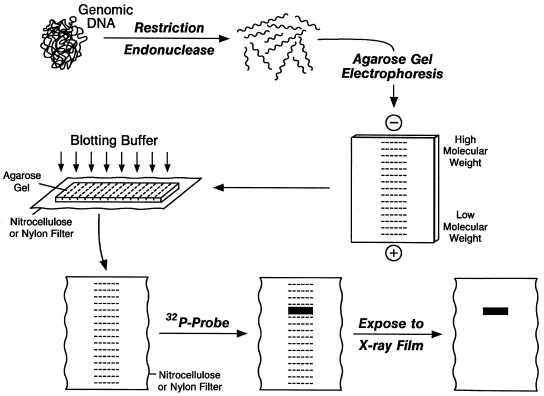
Southern blotting
- Develop by. E.M. Southern (1975).
- Technically, blotting may be defined as the transfer of macromolecules from the gel onto the surface of an immobilizing membrane like nitrocellulose membrane.
- In this technique first of all, the sample DNA is digested with restriction enzymes to obtain fragments of different lengths. These differently sized DNA segments are then passed through Agarose Gel Electrophoresis for their separation based on their lengths.
- The gel so obtained with different bands of DNA fragments is placed on top of buffer saturated filter papers which act as a filter paper wick.
- Above gel is put a nitrocellulose filter and over nitrocellulose filter are placed many dry filter paper sheets.
- With the movement of buffer towards the dry filter papers, the DNA bands are also moved upwards and hence they get bound to the nitrocellulose filter membrane.
- Now, the nitrocellulose filter is removed and baked in vacuum. DNA fragments on the nitrocellulose filter are hybridized with single stranded radioactively labeled probes.
- Washing is done to remove unbound probes and finally the DNA bands with radioactivity are visualized by autoradiography.
Northern Blotting
- In Northern Blotting, RNA molecules are blot transferred from the gel onto a chemically reactive paper.
Western blotting
- Western blotting is used for proteins and its working is based on the specificity of antibody-antigen reaction. In this technique the hybridization of bound proteins is done with radioactively labeled antibodies.
Dot Blot Hybridization:
- The procedure of this technique is almost the same as blotting, but the only difference is that the DNA fragments are not separated by electrophoresis, instead they are directly applied as a dot on the nitrocellulose membrane.
- Then radioactively labeled DNA probes having the complementary base sequences to the DNA of interest are applied on this membrane to allow its hybridization.
- The position of this hybridization is then detected by autoradiography method.
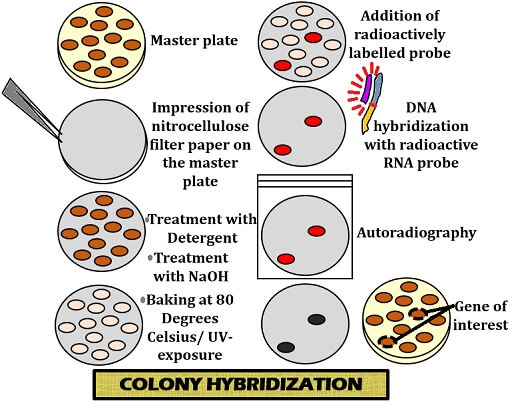
Colony Hybridization Technique:
- This technique is used in genetic engineering for the identification of transformed bacterial cells (i.e. cells which contain foreign DNA).
- After transformation of cells with a specific DNA, it is likely that only some of those cells may have foreign DNA. For further procedure, firstly it is important to screen such cells which are having foreign DNA.
- This screening is done by using the technique of colony hybridization in case of bacterial cells
- In this the transformed bacterial cells are first of all plated on a suitable agar plate which is termed as the master plate.
- Colonies are grown in the master plate. These colonies on the master plate are replica-plated onto a nitrocellulose or nylon membrane by placing it gently over the master plate.
- This replica-plate carrying the colonies is removed and treated with alkaline reagent to lyse the bacteria.
- DNA of those bacterial cells is denatured.
- Proteins on the membrane are digested.
- Finally the membrane is washed to remove all other molecules, leaving behind only the denatured DNA bound to it, in the form of DNA print of the colonies.
- This DNA print is then hybridized with a radioactively labeled RNA/DNA probe.
- Membrane is washed to remove any unbound probe and then autoradiography is done to detect radioactivity.
- The positions of the DNA prints showing up in autoradiograph are then compared with the master plate to identify the transformed colony.
Isolation of Recombinant Cells
- The transformation process generates a mixed population of transformed and non-trans- formed host cells.
- The selection process involves filtering the transformed host cells only.
- For isolation of recombinant cell from non-recombinant cell, marker gene of plasmid vector is employed.
- For examples, PBR322 plasmid vector contains different marker gene (Ampicillin resistant gene and Tetracycline resistant gene. When pst1 RE is used it knock out Ampicillin resistant gene from the plasmid, so that the recombinant cell become sensitive to Ampicillin.
Application of Recombinant DNA technology
- Recombinant DNA is widely used in biotechnology, medicine and research.
- The most common application of recombinant DNA is in basic research, in which the technology is important to most current work in the biological and biomedical sciences.
- Recombinant DNA is used to identify, map and sequence genes, and to determine their function.
- Recombinant proteins are widely used as reagents in laboratory experiments and to generate antibody probes for examining protein synthesis within cells and organisms.
- Many additional practical applications of recombinant DNA are found in industry, food production, human and veterinary medicine, agriculture, and bioengineering.
- DNA technology is also used to detect the presence of HIV in a person.
- Application of recombinant DNA technology in Agriculture – For example, manufacture of Bt-Cotton to protect the plant against ball worms.
- Application of medicines – Insulin production by DNA recombinant technology is a classic example.
- Gene Therapy – It is used as an attempt to correct the gene defects which give rise to heredity diseases.
- Clinical diagnosis – ELISA is an example where the application of recombinant DNA is possible.
Limitations of Recombinant DNA technology
- Destruction of native species in the environment the genetically modified species are introduced in.
- Resilient plants can theoretically give rise to resilient weeds which can be difficult to control.
- Cross contamination and migration of proprietary DNA between organisms.
- Recombinant organisms contaminating the natural environment.
- The recombinant organisms are population of clones, vulnerable in exact same ways. A single disease or pest can wipe out the entire population quickly.
- Creation of superbug is hypothesized.
- Ethical concern about humans trying to play God and mess with the nature’s way of selection. It is exaggerated by the fear of unknown of what all can be created using the technology and how is it going to impact the civilization.
- Such a system might lead to people having their genetic information stolen and used without permission.
- Many people worry about the safety of modifying food and medicines using recombinant DNA technology.

Related posts:
 Protoplast culture techniques
Protoplast culture techniques
 Selection of Somatic Hybrids
Selection of Somatic Hybrids
 Protoplast Fusion and Somatic Hybridization
Protoplast Fusion and Somatic Hybridization
 Isolation of protoplasts
Isolation of protoplasts
 DNA Fingerprinting
DNA Fingerprinting
 Identification of hybrid (cell) plants
Identification of hybrid (cell) plants
 Clonal Propagation (Micro propagation)
Clonal Propagation (Micro propagation)
 Gene Cloning Principle and Technique
Gene Cloning Principle and Technique
 Protoplast Regeneration
Protoplast Regeneration
 Limitation of Somatic Hybridization
Limitation of Somatic Hybridization


