Proto-oncogenes & Oncogenes
Proto-oncogenes
- A Retrovirus Can Transform a Host Cell by Inserting Its DNA Next to a Proto-oncogene of the Host.
- There are two ways in which a proto-oncogene can be converted into an oncogene upon incorporation into a retrovirus:
- the gene sequence may be altered or truncated so that it codes for a protein with abnormal activity, or the gene may be brought under the control of powerful promoters and enhancers in the viral genome that cause its product to be made in excess or in inappropriate circumstances.
- Retroviruses can also exert similar oncogenic effects in a different way:
- DNA copies of the viral RNA may simply be inserted into the host cell genome at sites close to, or even within, proto-oncogenes.
- The resulting genetic disruption is called an insertional mutation, and the altered genome is inherited by all the progeny of the original host cell.
- More or less random insertion of DNA copies of the viral RNA into the host DNA occurs as part of the normal retroviral life cycle, and in at least one well-documented case, insertion anywhere within about 10,000 nucleotide pairs from a proto-oncogene can cause abnormal activation of that gene.
- Insertional mutagenesis provides an important means of identifying proto-oncogenes, which can be tracked down by their proximity to the inserted viral DNA.
- Proto-oncogenes identified in this way often turn out to be the same as those discovered in the other way, as counterparts to oncogenes that retroviruses carry from cell to cell, but some new ones have been discovered as well.
- An example is the Wnt-1 gene, activated by insertional mutagenesis in breast cancers in mice infected with the mouse mammary tumor virus.
- This gene turns out to be closely homologous to the Drosophila gene wingless, which is involved in cell-cell communications that regulate details of the body pattern of the fly.

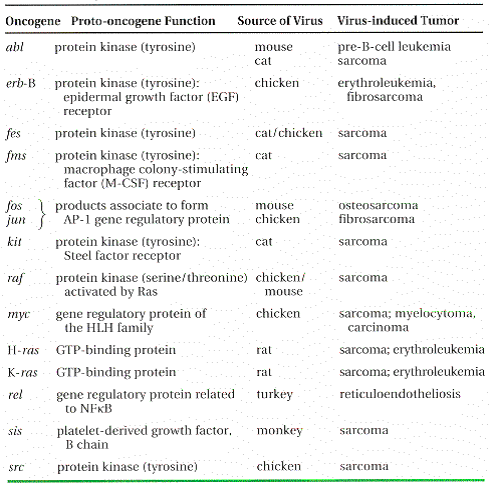
Table -Some Oncogenes Originally Identified Through Their Presence in Transforming Retroviruses
- Different Searches for the Genetic Basis of Cancer Converge on Disturbances in the Same Proto-oncogenes
- While some researchers pursued the line of investigation leading from retroviruses to oncogenes, others took a more direct approach and searched for DNA sequences in human cancer cells that would provoke uncontrolled proliferation when introduced into non cancerous cells.
- The assay was again done in cell culture, using an established line of mouse-derived fibroblast cells – NIH 3T3 cells – as the noncancerous hosts and transfecting them with DNA taken from human tumor cells.
- The findings were dramatic.
- Oncogenes were detected in many lines of human cancer cells, and in several cases these oncogenes turned out to be mutant alleles of some of the same proto-oncogenes that had been identified by the retroviral approach or of genes very closely related to them.
- About one in four human tumors, for example, was found to contain a mutated member of the ras gene family, first discovered as oncogenes carried by retroviruses that cause sarcomas in rats.
- Thus two independent lines of inquiry converged on the same genes.
- Yet another approach that led to some of the same proto-oncogenes was based on the karyotyping of tumor cells.
- As mentioned earlier, in almost all patients with chronic myelogenous leukemia, the leukemic cells show the same chromosomal translocation, between chromosomes 9 and 22; likewise, in Burkitt’s lymphoma there is regularly a translocation between chromosome 8 and one of the three chromosomes containing the genes that encode antibody molecules.
- In both these types of cancer the translocation breakpoint, where part of one chromosome is joined to another, was found to coincide exactly with the location of a proto-oncogene already known from retroviral studies – abl in chronic myelogenous leukemia, myc in Burkitt’s lymphoma.
- Analogous chromosome translocations are similarly associated with some other types of cancer.
- From DNA sequencing studies it seems that in some cases the translocation turns a proto-oncogene into an oncogene by fusing the proto-oncogene to another gene in such a way that an altered protein is produced in other cases the translocation moves a proto-oncogene into an inappropriate chromosomal environment that activates its transcription so that the normal protein is produced in excess.
- A Proto-oncogene Can Be Made Oncogenic in Many Ways – So far, about 60 proto-oncogenes have been discovered (Tables 3.1 and 3.2 show a small selection); each of these can be converted into an oncogene that plays a dominant part in cancers of one sort or another.
- Most such genes have been encountered repeatedly, in a variety of mutant forms and in several kinds of cancer, suggesting that the majority of mammalian proto-oncogenes may already have been identified.
Table 3.2. Some Oncogenes Originally Identified by Means Other Than Their Presence inTransforming Retroviruses
| S.N. | Means of Detection Oncogenes | |
| 1 | Insertional mutation Wnt-1 (int-1), fgf-3 (int-2), Notch-1 (int-3), lck | |
| 2 | Amplification L- myc, N- myc | |
| 3 | Transfection neu, N- ras, trk, ret | |
| 4 | Translocation bcl-2, RARa |
- But what functions do these genes have in a normal healthy cell, that mutations in them should be so dangerous?
- Most proto-oncogenes code for components of the mechanisms that regulate the social behavior of cells in the body – in particular, the mechanisms by which signals from a cell’s neighbors can impel it to divide, differentiate, or die.
- In fact, many of the components of cell signaling pathways were first identified through searches for oncogenes, and a full list of proto-oncogene products includes examples of practically every type of molecule involved in cell signaling – secreted proteins, transmembrane receptors, GTP-binding proteins, protein kinases, gene regulatory proteins, and so on, as summarized in Figure.
- All these molecules normally serve in complex relay chains to deliver signals for the production of more cells when more cells are needed.
- But mutations can alter them so that they deliver the signals even when more cells are not needed.
- The proto-oncogene erbB, for example, codes for the receptor for epidermal growth factor (EGF); when EGF binds to the receptor’s extra-cellular domain, the intracellular domain generates a stimulatory signal inside the cell.
- A mutation in c-erbB can turn it into an oncogene by deleting the extracellular EGF-binding domain in such a way that the intracellular stimulatory signal is produced constantly, even if no EGF is present.
- In a similar way a point mutation at an appropriate site in a ras gene can create a Ras protein that fails to hydrolyze its bound GTP and so persists abnormally in its active state, transmitting an intracellular signal for cell proliferation even when it should not. Innumerable other examples can be given.
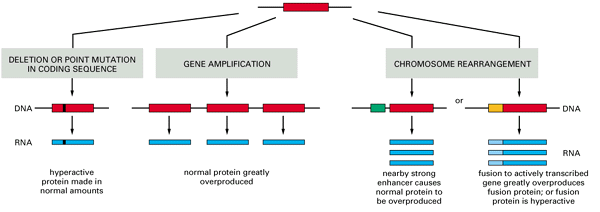
- The basic types of genetic accident that can convert a proto-oncogene into an oncogene are summarized in Figure .
- The gene may be altered by a point mutation, by a deletion, through a chromosomal translocation, or by insertion of a mobile genetic element such as retroviral DNA.
- The change can occur in the protein-coding region so as to yield a hyperactive product, or it can occur in adjacent control regions so that the gene is simply over expressed.
- Alternatively, the gene may be over expressed because it has been amplified to a high copy number through errors in the process of chromosome replication.

- Evidence from X-inactivation mosaics demonstrates the monoclonal origin of cancers.
- As a result of a random process that occurs in the early embryo, practically every normal tissue in a woman’s body is a mixture of cells with different X chromosomes heritably inactivated (indicated here by the mixture of red cells and gray cells in the normal tissue).
- When the cells of a cancer are tested for their expression of an X-linked marker gene, however, they are usually all found to have the same X chromosome inactivated.
- This implies that they are all derived from a single cancerous founder cell.
- Specific types of abnormality are characteristic of particular genes and of the responses to particular carcinogens.
- For example, 90% of the skin tumors evoked in mice by the tumor initiator dimethylbenz[a]anthracene (DMBA) have an A-to-T alteration at exactly the same site in a mutant ras gene; presume-ably, of the mutations caused by DMBA, it is only the ones at this site that efficiently activate skin cells to form a tumor.
- Members of the Myc gene family, on the other hand, are frequently over expressed or amplified.
- The Myc protein normally acts in the nucleus as a signal for cell proliferation, as excessive quantities of Myc cause the cell to embark on the cell-division cycle in circumstances where a normal cell would halt.
Oncogenes
- An oncogene is a gene that when mutated or expressed at abnormally-high levels contributes to converting a normal cell into a cancer cell.
- Cancer cells are cells that are engaged in uncontrolled mitosis.
- The signals for normal mitosis Normal cells growing in culture will not divide unless they are stimulated by one or more growth factors present in the culture medium.
- Example: PDGF (platelet-derived growth factor), which is encoded by the gene PDGFB (also known as SIS).
- The molecules of growth factor bind to molecules of its receptor, an integral membrane protein embedded in the plasma membrane with its ligand-binding site exposed at the surface of the cell. Example: the protein encoded by the gene ERBB2 encodes a receptor for epidermal growth factor (EGF). (In humans, ERBB2 is also known as HER2.)
- Binding of a growth factor to its receptor triggers a cascade of signaling events within the cytosol.
- Many of these involve kinases — enzymes that attach phosphate groups to other proteins. Examples: the proteins encoded by SRC, RAF, ABL, and the fusion protein encoded by BCR/ABL found in chronic myelogenous leukemia (CML).
- Or molecules that turn on kinases. Example: RAS. RAS molecules reside on the inner surface of the plasma membrane where they serve to link receptor activation to “downstream” kinases like RAF.
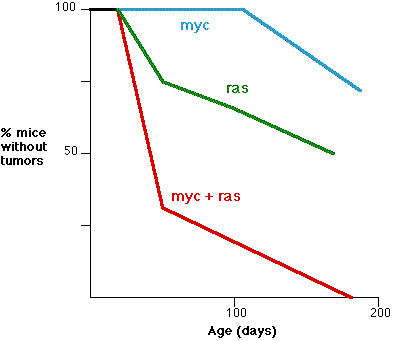
- This graph (based on the work of E. Sinn et al, Cell 49:465,1987) shows the synergistic effect of two oncogenes. The fraction (%) of transgenic mice without tumors is shown as a function of age.
- Three groups are shown: those mice transgenic for a hyperactive myc alone (blue) those transgenic for ras alone (green) those transgenic for both myc and ras (red) In most cases, phosphorylation activates the protein and eventually transfers the signal into the nucleus.
- Here phosphorylation activates transcription factors that bind to promoters and enhancers in DNA, turning on their associated genes. Examples: AP-1, a heterodimer of the proteins encoded by jun and fos.
- Some of the genes turned on by these transcription factors encode other transcription factors. Example: myc.
- Some of the genes turned on by these downstream transcription factors encode cyclins that prepare the cell to undergo mitosis.
- Genes that participate in any one of the steps above can become oncogenes if:
- they become mutated so that their product becomes constitutively active (that is, active all the time even in the absence of a positive signal) or
- they produce their product in excess. Possible causes:
- Their promoter and/or enhancer have become mutated.
- Example: the oncomouse: a transgenic mouse that has both copies of its myc gene under the influence of extra-powerful promoters.
- Loss (e.g., by a translocation) of the 3′ UTR of their mRNA so that a microRNA (miRNA) that normally represses translation can no longer do so.
- All these oncogenes act as dominants; if the cell has one normal gene (sometimes called a proto-oncogene) at a locus and one mutated gene (the oncogene), the abnormal product takes control.
- No single oncogene can, by itself, cause cancer. It can, however, increase the rate of mitosis of the cell in which it finds itself.
- Dividing cells are at increased risk of acquiring mutations, so a clone of actively dividing cells can yield subclones of cells with a second, third, etc. oncogene.
- When a clone loses all control over its mitosis it is well on its way to developing into a cancer.
Other types of potential cancer-promoting genes –
- Genes that inhibit apoptosis.
- The suicide of damaged cells (apoptosis) provides an important mechanism for ridding the body of cells that could go on to form a cancer.
- It is not surprising then that inhibiting apoptosis can promote the formation of a cancer.
- Example: Bcl-2. The product of this gene inhibits apoptosis. Over expression of the gene is a hallmark of B-cell cancers.
Genes involved in repairing DNA or stopping mitosis if they fail –
- Mutations arise from an unrepaired error in DNA. So any gene whose product participates in DNA repair probably can also behave as an oncogene when mutated.
- Example: ATM. ATM (=”ataxia telangiectasia mutated”) gets its name from a human disease of that name, whose patients – among other things – are at increased risk of cancer.
- The ATM protein is also involved in detecting DNA damage and interrupting the cell cycle when damage is found.
- It is estimated that fully 1% of the 25,000 or so genes in the human genome are proto-oncogenes.
Tumor-Suppressor Genes
- The products of some genes inhibit mitosis. These genes are called tumor suppressor genes.

Related posts:
 DNA Damage Repair Mechanism & Defect On Repair Mechanism
DNA Damage Repair Mechanism & Defect On Repair Mechanism
 Sex Limited Characters and sex reversal
Sex Limited Characters and sex reversal
 MUTATIONS -Spontaneous & Induced Mutations
MUTATIONS -Spontaneous & Induced Mutations
 Chemical and Physical Mutagens
Chemical and Physical Mutagens
 EUPLOIDY
EUPLOIDY
 Polyploidy
Polyploidy
 Multiple Alleles
Multiple Alleles


