Strategies of Genetic Engineering:
According to the requirement there are different routes of genetic engineering and a person prefers as per need and choice. Fig. 11.1 gives a general outline of DNA cloning.
The overall strategies involve the major four steps:
(i) Formation of DNA fragments,
(ii) Joining of DNA into vectors,
(iii) Introduction of vectors into host cells, and
(iv) Selection of newly acquired DNA.

Formation of DNA Fragments:
- The DNA fragments to be cloned are called foreign DNA or passenger DNA. The DNA fragment of known function is selected and identified. It is isolated from the organism by several in vitro biochemical methods. In addition, the DNA fragments can be constructed chemically by using mRNA of gene machine.
- By using mRNA, the complementary DNA (cDNA) molecules are produced. All these processes are possible only due to enzymes, the nucleases, DNA polymerases, DNA ligases and restriction endonucleases or restriction enzymes.
- a. Restriction Enzymes:
- Restriction enzymes occur in bacteria as a chemical weapon against the invading viruses and cut both the strands of DNA when certain foreign nucleotides are introduced in a bacterial cell. Now a days, many restriction enzymes are known.
- The first restriction enzyme was isolated in 1970 from Haemophilus influenzae. However, different restriction enzymes present in different bacteria recognize different nucleotide sequences. The restriction enzymes cleave a DNA to generate a nick with a 5′ phosphoryl and 3′ OH termini. The broken nucleotides form a DNA duplex and exhibit two fold symmetry from a point.
- In some cases cleavage in two strands are staggered to produce single strand short projections opposite to each other with blunt ends of mutually cohesive stickily ends which are identical and complementary sequences are called palindrome sequences or palindromes.
- Therefore, when read from 5’→3′, both strands have the same sequence. Now-a-days, a large number of restriction enzymes are commercially available. Some of the commonly used restriction enzymes are given in Table 11.1.
Table 11. 1 : Source of restriction enzyme, cleavage site and products of cleavage.

Arrows indicate the recognition of restriction sites.
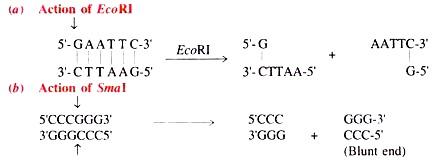
(i) Action of Restriction Enzymes:
The restriction enzymes recognise the cleavage sites and result in cohesive ends as shown in Table given above. =
B. Use of Linkers, Adaptors and Homopolymer Tails:
- Some times after use of restriction enzymes there is no generation of stickily ends. DNA molecule is fragmented that contains blunt ends as above. In such situation, the recombination frequency lowers due to non-availability of suitable sites on the DNA to be manipulated. Hence, cleavage sites can be added as linkers, adaptor molecules or homopolymer tails.
- Linkers are the chemically synthesized dsDNA oligonucleotides containing on it one or more cleavage sites for restriction enzymes. Linkers are ligated to the blunt ends of DNA to be cloned by using T4 DNA ligase. Then these are cut with specific restriction enzyme to generate DNA fragments with sticky ends (Fig. 11.2A).

- The adaptor molecules are the chemically synthesized DNA molecules with preformed cohesive ends. The adaptors are used when the target cleavage site for the restriction enzyme used is located within the DNA sequence (i.e. foreign DNA) to be cloned (Fig.11.2B).
- The homopolymers are oligo dA (AAA………… A) Sequences and oligo dT (TTT….T) sequences. These sequences are added to the foreign DNA fragments to be cloned and vector used by terminal transferase enzyme.
- The homopolymers are used in such a way that if foreign DNA is to add oligo dA, the vector will be added with oligo dT. Consequently, there develops oligo dA and oligo dT tails on foreign DNA and vector, respectively. When mixed together they anneal to form the covalently closed circular rDNA molecules (Fig. 11.3).

ii. Joining of DNA into Vectors:
- The small sequences of DNA can be spliced or joined into the vector DNA by an enzyme, DNA ligase resulting into the creation of artificial DNA vector i.e. recombinant vectors.
- a. DNA Ligases:
- DNA ligases seal the cut ends of two DNA molecules.
- b. The Cloning Vectors:
- Vectors are those DNA molecules that can carry a foreign DNA fragment when inserted into vectors. These are also called as vehicle DNA because they act as carrier of genes to be cloned into a recipient cell. There are many types of vectors used in genetic engineering experiments such as plasmids, bacteriophages, cosmids, phasmids and shuttle vectors.
- Moreover, vectors can be introduced into the host cell, thereafter they replicate independently by using the host enzymes. The cloned (inserted) foreign DNA segment can be amplified along the vector inside the host cell.
- (i) Plasmid Vectors:
- Plasmids are the extra chromosomal, self replicating and double stranded closed and circular DNA molecules present in the bacterial cell. They contain several genetic information’s for their own replication. They specify a number of host properties. On the basis of these characters they have been grouped into several types.
- Plasmids can be considered a suitable cloning vector if they bear the following features:
- (a) They can be isolated from the cells but not found naturally.
(b) They must possess at least one cleavage site for one or more restriction enzymes.
(c) Insertion of a linear molecule at one of these sites does not alter its replication properties.
(d) They can be introduced into a bacterial cell and cells carrying plasmid with or without insert can be selected or identified.
- However, the conjugative (self-transmissible) plasmids carry transfer (tra) genes that enable them to transfer from one bacterium to the another. Non- conjugative plasmids do not carry tra genes. They replicate autonomously but cannot transfer to another bacterium. In contrast, the relaxed plasmids are found as multiple copies in a cell. Plasmids present typically in 1-2 copies per cell are called stringent plasmids.
- Moreover, the number of plasmids in a bacterial cell can be increased to about 1000 per cell. This process of increasing the number of plasmids is called amplification. Number of plasmids can be amplified by incubation of the host cells with the antibiotic chloramphenicol. It inhibits the proteins required for replication of chromosome but does not inhibit plasmid replication.
- Some of the important plasmids are given in Table 11.2. A physical map of plasmid pBR322 is shown in Fig. 11.4. Plasmid pBR322 is one of the most widely used multi-copy cloning vectors of E. coli and is a hybrid vector of COI EL and genes coding for resistance against tetracycline and ampicillin.
- Plasmid pBR322 consists of genes for origin of replication (ori) for resistance to tetracycline (tetr) and ampicillin (ampr), unique recognition for 20 restriction enzymes.

- Different types of plasmids of E. coli and Bacillus are listed in Table 11.2A-C. Different species of Pseudomonas, Streptococcus, Salmonella, etc. possess a variety of plasmids of different size and markers.
(ii) Bacteriophage Vectors:
- The filamentous bacteriophages are M13, Fd and F1 that contain single stranded circular DNA molecule. They are used for cloning of DNA fragments. Examples of phage vectors are given in Table. 11.3. In addition, bacteriophage λ is a phage of E. coli and contains double stranded DNA molecule of about 49.5 kilo base pairs (kb).
- The ssDNA exists as a linear molecule that consists of 12 nucleotides long single stranded 5′ ends. However, there is possibility to introduce the foreign DNA up to 25 kb in length into the lambda genome without making them incapable to infect E. coli and replicate properly.

- Bacteriophage λ consists of several non-essential regions which can be replaced with almost equal length of foreign DNA molecule to be cloned.
However, there are certain advantages of phage cloning vectors over the plasmids:
(a) DNA can be packed into the phage particles and transduced into E. coli with high efficiency.
(b) The upper size limit of insertion of foreign DNA into the phage is about 35 kb in length.
(c) Screening and storage of recombinant DNA is easier in phage λ than plasmids.
- Two types of cloning vectors have been constructed: insertion vector and replacement vector. Insertion vectors are those in which relatively a fragment of DNA of short length is inserted without removal of any portion of phage DNA.
- The replacement vectors are those in which case first the non-essential region is cut and removed and almost equal length of foreign DNA is inserted. Thus, the foreign DNA replaces the non-essential region of phage λ. For detail see A Textbook of Biotechnology by R.C. Dubey.
- Phage λ DNA has been widely used as cloning vector because of ease in handling and screening of a large number of recombinant DNA containing phages. That is why it is used as a biological tool in building the gene bank, for example rat, yeast, mouse and human gene bank.
(iii) Cosmid Vectors:
- Based on the properties of phage DNA and Col El plasmid DNA, a group of Japanese workers showed that the presence of a small segment of phage λ containing cohesive end (cos site) on the plasmid DNA molecule is a sufficient prerequisite for in vitro packaging of this DNA into the infection particles.
- Therefore, the cosmids are the hybrid vectors derived from plasmids and phage λ (only cos site). Cosmid was constructed for the first time by Collins and Hohn (1978).
- Examples of cosmid vectors are given in Table11.3:

- Cosmids lack genes coding for viral proteins, therefore, neither viral particles are formed nor cell lysis occurs. The special features of cosmids similar to plasmids are the presence of origin of replication, markers (conferring resistance against antibiotics), a special cleavage site for the insertion of foreign DNA, and their small size.
- Phage λ consists of 49.5 kb long DNA in which cos site has 12 base pairs. The cos site is ligated to 5 kb pairs long plasmid containing markers. Therefore, it is possible to insert about 45 kb long foreign DNA, which is larger than it would be possible to be done in phage X or plasmid vector.
- Procedure of DNA cloning by using cosmid vector is shown in Fig. 11.5. After infection, the DNA of cosmid vector cyclizes at cos site and replicates as plasmid and expresses the drug resistance markers. In recent years, a number of cosmid vectors has been constructed from E. coli, yeast and mammalian cells and gene bank has been built up.

(iv) Phagemid Cloning Vectors:
- Like cosmids, phagemids are also the hybrids of a plasmid and a phage. The plasmids are restricted to intracellular state, while the phage particles can exist extra-cellularly as the infection particles. Kahn and Helinski (1978) reconstructed the plasmid of Col El artificially and allowed to get packed in vitro into bacteriophage particles. The phage particles containing plasmid DNA were allowed to infect bacterium.
- Thus the hybrid vector was termed as phagemids. This insertion of plasmid into phage genome is reversible and called as lifting’ the plasmid. It generates a phage genome containing att site and one or more plasmid site(s).
- These new genetic re-combinations called as phagemids. The phagemids contains functional ori genes of plagemid and of phage λ. Moreover, they may be allowed to propagate as plasmid of phage in appropriate E. coli strains. After reversal of lifting process plasmids are released.
(v) Shuttle Vector:
- The shuttle vectors are the plasmids that are designated to replicate in different host systems i.e. in prokaryotes and eukaryotes. A shuttle vector is constructed by using bacterial origin of replication in a yeast plasmid. Thus the origins of replication of different host systems such as E. coli or yeast are combined in one plasmid.
- This is why any gene inserted into shuttle vector can be expressed either in bacterium E. coli or Bacillus or yeast cells. Therefore, shuttle vectors transform E. coli cells with greater efficiency than the original organisms. For example, a prokaryotic gene for β-lactamase is expressed in E. coli.
- Genetic map of a shuttle vector constructed by E. coli plasmid and yeast is shown in Fig. 11.6. The important gene loci are ars (autonomously replicating sequence), cen (centromere of yeast), leu-2 (complements of a defective gene encoding for leucin, ori (origin for replication in prokaryotes) and ampr (ampicillin resistance).
c. Insertion of Foreign DNA into a Vector:
- The foreign DNA of desired function to be cloned is either synthesized artificially following chemical methods or gene machine or synthesized by using mRNA of known function. The cDNA can also be procured from cDNA bank also.
- The vectors are used as desired. Both the vector and foreign DNA are treated with restriction enzyme to generate cohesive (slickly) ends of identical base pairs (Fig. 11.7A-B). Both the DNA molecules are mixed together with ATP and T4 DNA ligase which forms phosphodiester bonds between the stickily ends and thus permanently seal them (C).
- Thus a hybrid of foreign DNA molecule and vector is obtained which is called recombinant DNA or rDNA. The rDNA is now circularized. In order to get efficient rDNA molecules addition of cohesive ends on both termini of foreign DNA and vector DNA is necessary. There are three ways of generating efficient rDNA molecules (see linkers, adaptors and homopolymer tails).
iii. Introduction of Recombinant Vector into the Recipient Cells (transformation):
- After preparing the rDNA it is allowed to enter into a suitable host cell for expression of foreign DNA. Originally, the procedure of transformation of bacterial cells by rDNA of vector was developed by Mandell and Higa (1979).
- The strains of E. coli possess restriction enzymes, hence they degrade foreign DNA. To escape from degradation, the exponentially growing cells are pre-treated with CaCl2 at low temperature; thereafter, the vector DNA is mixed up. Depending upon time, the vector DNA enters the bacterial cell. This process is called transformation (Fig.11.7C-D).

- For the first time phage X was used to transfer the foreign DNA into E. coli cells. Thus, the process of transfer of DNA by a phage virus is called transfection similar to transduction.
- Expression of Cloned Genes (in E. coli)
- There are many factors that influence the expression of a cloned gene in E. coli.
Some of them are discussed below:
(i) Commonly vectors are constructed such that the DNA insert is flanked by promoters for two different viral RNA polymerases of phage T3, T7 or SP6. Thus efficiency of promoters influences the expression of cloned gene.
(ii) The foreign gene of interest should be cloned into a high copy number plasmid such as pBR322. Replication of pBR322 is controlled by an RNA molecule called RNA I. It binds
to a second RNA molecule called RNA II and, therefore, inhibits replication of chromosomal DNA.
- However, sometimes copy number is controlled by promoters whose activity is regulated by changing the temperature. Such vectors are called runaway plasmid vectors. After increasing the temperature from 30°C to 38°C control of plasmid over replication is lost. This results in increase in copy number of plasmid.
(iii) Upstream to initiation codon (AUG) the sequences are known to influence the translational efficiency. This is called Shine Dalgamo (SD) sequence (5′ UAAGGAGGU3′) which acts as a ribosome binding site. While constructing a plasmid, presence of a SD sequence should be kept in mind.
iv. Selection of Clones:
- Not all, but some of the transformed bacterial cells contain foreign gene where it functions properly. Such cells are selected from the mass of cells by using antibiotics.
- For example the plasmids of rDNA so obtained (Fig. 11.7 C) are introduced in E. coli cells. The transformed cells are plated onto nutrient medium containing ampicillin. The antibiotic marker gene (ampr) present on pBR322 determines resistance to ampicillln. Therefore, such cells will grow on medium and form colonies. This is called as master plate.
- To ascertain the presence or absence of tetr gene in the cloned plasmid, a replica plating from the master plate is done. Replica plates are incubated for the growth of bacterial colonies.
- The appearance of colonies on replica plates is compared with the master plate and those colonies that fail to grow on replica plate are supposed to have plasmids where tetr gene is destroyed while those growing on replica plate show that both genes (ampr and tetr) are present in plasmid.
- Moreover, the foreign DNA cloned in plasmid vector is now functioning or not is also to be tested and selected from thousands of cells. There are several approaches, which ascertain about the presence and functioning of foreign DNA into transformed cells. Foreign DNA of known function transferred through vector in E. coli cells can be observed by colony hybridization (nucleic acid hybridization) method, in vitro translation method and immunological tests.
- a. Colony Hybridization (Nucleic Acid Hybridization) Technique:
- The colony hybridization technique has been given by Grustein and Hogness (1975). It is based on the availability of radioactively labelled DNA probe. A probe is a radioactively labelled (P32) oligonucleotide (20-10 nucleotides long) with a sequence complementary to at least one part of the desired DNA.
- The probe may be even partially pure mRNA of related gene that defects corresponding rDNA. DNA probes have commercial significance in disease diagnosis, finger printing, microbiological tests and research as well.
This technique follows replica plating of bacterial colonies on nitrocellulose filter disc (Fig. 11.8 A) which is then placed on gelled nutrient medium and both master plate and disc are incubated to grow the bacterial colonies.
Colonies growing on nitrocellulose filter paper derive nutrients from gelled medium through pores
(B). The filter paper is removed and placed on blotting paper soaked with 0.5 N NaOH solutions. The alkali lyses bacteria and denatures their DNA.
- Then the disc is neutralized by tris (hydroxymethyl) amino methane -HCl buffer by maintaining high concentrations of the salt. Thus, the DNA binds to the disc in the same pattern as the bacterial colonies did. The filter disc is baked at 80°C so that DNA may fix properly (C).

- The disc is incubated with a solution containing radioactively labelled probe (P32) xhe probe hybridizes any bound DNA that contains sequences complementary to the probe (D). Disc is washed thoroughly to remove un-hybridized probes and it is passed through X-rays (E-F). Colonies that develop positive X-ray image (G) are compared with master plate and picked up and multiplied in nutrient medium.
B. Plaque Hybridization Technique:
Plaque hybridization technique is analogous to colony hybridization technique. It has been given for phage.
Similarly this technique follows:
(i) Cleavage of cellular DNA and bacteriophage DNA separately,
(ii) Ligation of foreign DNA fragments randomly with phage DNA resulting in recombinant DNA,
(iii) Allowing the rDNA for packaging and producing hybrid phage artificially
(iv) Plating of recombinant phage with host bacteria (phage clones are produced on bacterial lawns),
(v) Isolation of phage clones and establishment of genomic library (each colony of the bacterial lawn is a recombinant clone carrying different DNA fragments),
(vi) Second stage plating of phage clones to be analysed with host bacteria,
(vii) Replica plating on nitrocellulose filter paper and incubation of filter paper on gelled medium,
(viii) Lysis of bacteria with alkali and neutralizing with HCl buffer,
(ix) Adding radiolabelled probe and passing of filter paper through X-ray,
(x) Detection of clone from master plate and selection of desired radiolabelled phage.
c. The Southern Blotting Technique:
- In 1975, Edwin M. Southern (a molecular biologist) published a procedure/or detecting specific DNA fragments so that a particular gene could be isolated from a complex DNA mixture. The procedure starts with digestion of DNA population by one or more restriction enzymes (Fig. 11.9). Consequently DNA fragments of unequal length are produced.
- This preparation is passed through agarose gel electrophoresis that results in separation of DNA molecules according to their respective size. The DNA fragments present in gel are denatured by alkali treatment. Then gel is put on top of buffer saturated with filter paper. Upper surface of gel is then covered with nitrocellulose filter and overlaid with dry filter paper. The dry filter paper draws the buffer through gel.
- Buffer contains single stranded DNA. Nitrocellulose filter paper binds DNA fragments strongly when comes in contact of it. After baking at 80°C, DNA fragments are permanently fixed to the nitrocellulose filter. Then the filter is placed in a solution containing radio-labelled DNA probe of known sequences for about 20 minutes. These are complementary in sequence to the blot transferred DNA.

- The radiolabelled nucleic acid probe hybridizes the complementary DNA on nitro cellulose in about 20 minutes. The filter is thoroughly washed to remove the probe. The hybridized regions are detected auto-radio-graphically by placing the nitrocellulose filter in contact with a photographic film. The images show the hybridized DNA molecules. Thus, the sequences of DNA are recognised following the sequences of nucleic acid probe.






