Inherited Human Disease Related with the Gene
Many human diseases are caused by defective genes. A few common examples:
| Disease | Genetic defect |
| hemophilia A | absence of clotting factor VIII |
| cystic fibrosis | defective chloride channel protein |
| muscular dystrophy | defective muscle protein (dystrophin) |
| sickle-cell disease | defective beta globin |
| severe combined immuno-deficiency (SCID) | any one of several genes fail to make a protein essential for T and B cell function |
- All of these diseases are caused by a defect at a single gene locus. (The inheritance is recessive so both the maternal and paternal copies of the gene must be defective.)
Hemophilia A –
- Hemophilia A is a hereditary blood disorder, primarily affecting males, characterized by a deficiency of the blood clotting protein known as Factor VIII that results in abnormal bleeding.
- Babylonian Jews first described hemophilia more than 1700 years ago; the disease first drew widespread public attention when Queen Victoria transmitted it to several European royal families.
- Mutation of the HEMA gene on the X chromosome causes Hemophilia A. Normally, females have two X chromosomes, whereas males have one X and one Y chromosome.
- Since males have only a single copy of any gene located on the X chromosome, they cannot offset damage to that gene with an additional copy as can females.
- Consequently, X-linked disorders such as Hemophilia A are far more common in males.
- The HEMA gene codes for Factor VIII, which is synthesized mainly in the liver, and is one of many factors involved in blood coagulation; its loss alone is enough to cause Hemophilia A even if all the other coagulation factors are still present.
- Treatment of Hemophilia A has progressed rapidly since the middle of the last century when patients were infused with plasma or processed plasma products to replace Factor VIII.
- HIV contamination of human blood supplies and the consequent HIV infection of most hemophiliacs in the mid-1980s forced the development of alternate Factor VIII sources for replacement therapy, including monoclonal antibody purified Factor VIII and recombinant Factor VIII, both of which are used in replacement therapies today.
- Hemophilia A has reached the clinical trial stage, and results so far have been encouraging.
- Investigators are still evaluating the long-term safety of these therapies, and it is hoped that a genetic cure for hemophilia will be generally available in the future.
Cystic fibrosis –
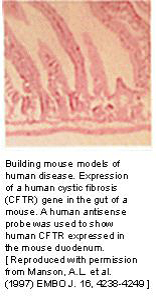
- Cystic fibrosis (CF) (Figure 3.40) is the most common fatal genetic disease in the United States today.
- It causes the body to produce a thick, sticky mucus that clogs the lungs, leading to infection, and blocks the pancreas, stopping digestive enzymes from reaching the intestines where they are required to digest food.
- CF is caused by a defective gene, which codes for a chloride transporter found on the surface of the epithelial cells that line the lungs and other organs.
- Several hundred mutations have been found in this gene, all of which result in defective transport of chloride, and secondarily sodium, by epithelial cells.
- As a result, the amount of sodium chloride (salt) is increased in bodily secretions.
- The severity of the disease symptoms of CF is directly related to the characteristic effects of the particular mutation(s) that have been inherited by the sufferer.
- CF research has accelerated sharply since the discovery of CFTR in 1989. In 1990, scientists successfully cloned the normal gene and added it to CF cells in the laboratory, which corrected the defective chloride transport mechanism.
- This technique – gene therapy – was then tried on a limited number of CF patients.
- However, this treatment may not be as successful as originally hoped.
- Further research will be required before gene therapy, and other experimental treatments, prove useful in combating CF.
Duchenne muscular dystrophy –
- Duchenne muscular dystrophy (DMD) is one of a group of muscular dystrophies characterized by the enlargement of muscles (Figure 3.41).
- DMD is one of the most prevalent types of muscular dystrophy and is characterized by rapid progression of muscle degeneration that occurs early in life.
- All are X-linked and affect mainly males – an estimated 1 in 3500 boys worldwide.
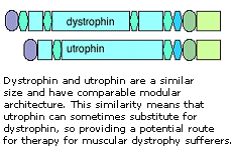
- The gene for DMD, found on the X chromosome, encodes a large protein – dystrophin.
- Dystrophin is required inside muscle cells for structural support; it is thought to strengthen muscle cells by anchoring elements of the internal cytoskeleton to the surface membrane.
- Without it, the cell membrane becomes permeable, so that extracellular components enter the cell, increasing the internal pressure until the muscle cell “explodes” and dies.
- The subsequent immune response can add to the damage.
- A mouse model for DMD exists and is proving useful for furthering our understanding on both the normal function of dystrophin and the pathology of the disease.
- In particular, initial experiments that increase the production of utrophin, a dystrophin relative, in order to compensate for the loss of dystrophin in the mouse are promising and may lead to the development of effective therapies for this devastating disease.
Sickle Cell Anemia –
- Sickle cell anemia (Figure 3.42) is the most common inherited blood disorder in the United States, affecting about 72,000 Americans or 1 in 500 African Americans.
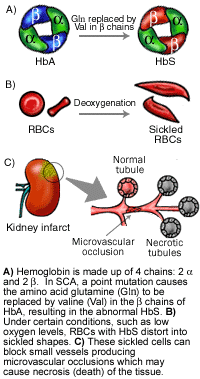
- SCA is characterized by episodes of pain, chronic hemolytic anemia and severe infections, usually beginning in early childhood. SCA is an autosomal recessive disease caused by a point mutation in the hemoglobin beta gene (HBB) found on chromosome 11p15.4.
- ]Carrier frequency of HBB varies significantly around the world, with high rates associated with zones of high malaria incidence, since carriers are somewhat protected against malaria. About 8% of the African American populations are carriers.
- A mutation in HBB results in the production of a structurally abnormal hemoglobin (Hb), called HbS. Hb is an oxygen carrying protein that gives red blood cells (RBC) their characteristic color.
- Under certain conditions, like low oxygen levels or high hemoglobin concentrations, in individuals who are homozygous for HbS, the abnormal HbS clusters together, distorting the RBCs into sickled shapes.
- These deformed and rigid RBCs become trapped within small blood vessels and block them, producing pain and eventually damaging organs.
- Though, as yet, there is no cure for SCA, a combination of fluids, painkillers, antibiotics and transfusions are used to treat symptoms and complications.
- Hydroxyurea, an antitumor drug, has been shown to be effective in preventing painful crises.
- Hydroxyurea induces the formation of fetal Hb (HbF) – a Hb normally found in the fetus or newborn— which, when present in individuals with SCA, prevents sickling.
- A mouse model of SCA has been developed and is being used to evaluate the effectiveness of potential new therapies for SCA.
Severe combined immunodeficiency –
- Severe combined immunodeficiency (SCID) represents a group of rare, sometimes fatal, congenital disorders characterized by little or no immune response (Figure 3.43).

- The defining feature of SCID, commonly known as “bubble boy” disease, is a defect in the specialized white blood cells (B- and T-lymphocytes) that defend us from infection by viruses, bacteria and fungi.
- Without a functional immune system, SCID patients are susceptible to recurrent infections such as pneumonia, meningitis and chicken pox, and can die before the first year of life.
- Though invasive, new treatments such as bone marrow and stem-cell transplantation save as many as 80% of SCID patients.
- All forms of SCID are inherited, with as many as half of SCID cases linked to the X chromosome, passed on by the mother. X-linked SCID results from a mutation in the interleukin 2 receptor gamma (IL2RG) gene which produces the common gamma chain subunit, a component of several IL receptors.
- IL2RG activates an important signalling molecule, JAK3. A mutation in JAK3, located on chromosome 19, can also result in SCID.
- Defective IL receptors and IL receptor pathways prevent the proper development of T-lymphocytes that play a key role in identifying invading agents as well as activating and regulating other cells of the immune system.
- In another form of SCID, there is a lack of the enzyme adenosine deaminase (ADA), coded for by a gene on chromosome 20.
- This means that the substrates for this enzyme accumulate in cells.
- Immature lymphoid cells of the immune system are particularly sensitive to the toxic effects of these unused substrates, so fail to reach maturity.
- As a result, the immune system of the afflicted individual is severely compromised or completely lacking.
- Some of the most promising developments in the search for new therapies for SCID center on ‘SCID mice’, which can be bred deficient in various genes including ADA, JAK3, and IL2RG.
- It is now possible to reconstitute the impaired mouse immune system by using human components, so these animals provide a very useful model for studying both normal and pathological immune systems in biomedical research.
Severe combined immunodeficiency (SCID) –
- SCID is a disease in which the patient has neither cell-mediated immune responses nor is able to make antibodies.
- It is a disease of young children because, until recently, the absence of an immune system left them prey to infections that ultimately killed them.
- About 25% of the cases of SCID are the result of the child being homozygous for defective genes encoding the enzyme adenosine deaminase (ADA).
- The normal catabolism of purines is deficient, and this is particularly toxic for T cells and B cells.
Thalassemia –
- Thalassemia is an inherited disease of faulty synthesis of hemoglobin (Figure 3.44).

- The name is derived from the Greek word “thalassa” meaning “the sea” because the condition was first described in populations living near the Mediterranean Sea; however, the disease is also prevalent in Africa, the Middle East, and Asia.
- Thalassemia consists of a group of disorders that may range from a barely detectable abnormality of blood, to severe or fatal anemia.
- Adult hemoglobin is composed of two alpha (α) and two beta (β) polypeptide chains.
- There are two copies of the hemoglobin alpha gene (HBA1 and HBA2), which each encode an α -chain, and both genes are located on chromosome 16.
- The hemoglobin beta gene (HBB) encodes the β-chain and is located on chromosome 11.
- In α-thalassemia, there is deficient synthesis of α-chains.
- The resulting excess of β-chains bind oxygen poorly, leading to a low concentration of oxygen in tissues (hypoxemia).
- Similarly, in β-thalassemia there is a lack of β-chains. However, the excess α-chains can form insoluble aggregates inside red blood cells.
- These aggregates cause the death of red blood cells and their precursors, causing a very severe anemia.
- The spleen becomes enlarged as it removes damaged red blood cells from the circulation.
- Deletions of HBA1 and/or HBA2 tend to underlie most cases of α-thalassemia.
- The severity of symptoms depends on how many of these genes are lost.
- Loss of one or two genes is usually asymptomatic, whereas deletion of all four genes is fatal to the unborn child.
- In contrast, over 100 types of mutations affect HBB, and deletion mutations are rare. Splice mutations and mutations that occur in the HBB gene promoter region tend to cause a reduction, rather than a complete absence, of β-globin chains and so result in milder disease.
- Nonsense mutations and frameshift mutations tend to not produce any β-globin chains leading to severe disease.
- Currently, severe thalassemia is treated by blood transfusions, and a minority of patients are cured by bone marrow transplantation.
- Mouse models are proving to be useful in assessing the potential of gene therapy.
Friedreich’s ataxia –
- Friedreich’s ataxia (FRDA) is a rare inherited disease characterized by the progressive loss of voluntary muscular coordination (ataxia) and heart enlargement.
- It is named after the German doctor, Nikolaus Friedreich, who first described the disease in 1863.
- FRDA is generally diagnosed in childhood and affects both males and females.
- FRDA is an autosomal recessive disease caused by a mutation of a gene called frataxin, which is located on chromosome 9 .

- This mutation means that there are many extra copies of a DNA segment, the trinucleotide GAA.
- A normal individual has 8 to 30 copies of this trinucleotide, while FRDA patients have as many as 1000.
- The larger the number of GAA copies, the earlier the onset of the disease and the quicker the decline of the patient.
- Although we know that frataxin is found in the mitochondria of humans, we do not yet know its function.
- However, there is a very similar protein in yeast, YFH1, which we know more about. YFH1 is involved in controlling iron levels and respiratory function.
- Since frataxin and YFH1 are so similar, studying YFH1 may help us understand the role of frataxin in FRDA.
Lesch-Nyhan syndrome –
- Lesch-Nyhan syndrome (LNS) is a rare inherited disease that disrupts the metabolism of the raw material of genes.
- These raw materials are called purines, and they are an essential part of DNA and RNA.
- The body can either make purines (de novo synthesis) or recycle them (the resalvage pathway).
- Many enzymes are involved in these pathways. When one of these enzymes is missing, a wide range of problems can occur.
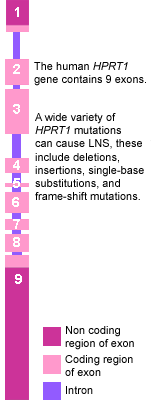
- In LNS, there is a mutation in the HPRT1 gene located on the X chromosome.
- The product of the normal gene is the enzyme hypoxanthine-guanine phosphoribosyltransfer-ase, which speeds up the recycling of purines from broken down DNA and RNA.
- Many different types of mutations affect this gene, and the result is a very low level of the enzyme.
- The mutation is inherited in an X-linked fashion. Females who inherit one copy of the mutation are not affected because they have two copies of the X chromosome (XX).
- Males are severely affected because they only have one X chromosome (XY), and therefore their only copy of the HPRT1 gene is mutated (Figure 3.46).
- Mutations of the HPRT1 gene cause three main problems.
- First is the accumulation of uric acid that normally would have been recycled into purines.
- Excess uric acid forms painful deposits in the skin (gout) and in the kidney and bladder (urate stones).
- The second problem is self-mutilation.
- Affected individuals have to be restrained from biting their fingers and tongues.
- Finally, there is mental retardation and severe muscle weakness.
- In the year 2000 it was shown that the genetic deficiency in LNS could be corrected in vitro.
- A virus was used to insert a normal copy of the HPRT1 gene into deficient human cells.
- Such techniques used in gene therapy may one day provide a cure for this disease.






