![]()
Mapping the Bacteriophage Genome
- A bacteriophage (from ‘bacteria’ and Greek ‘phagein= to eat’) is any one of a number of viruses that infect bacteria.
- The term is commonly used in its shortened form, phage.
- Typically, bacteriophages consist of an outer protein hull enclosing genetic material.
- The genetic material can be ssRNA (single stranded RNA), dsRNA, ssDNA, or dsDNA between 5 and 500 kilo base pairs long with either circular or linear arrangement.
- Bacteriophages are much smaller than the bacteria they destroy – usually between 20 and 200 nm in size.
- T2 and its close relative T4 are viruses that infect the bacterium E. coli.
- The infection ends with destruction (lysis) of the bacterial cell so these viruses are examples of bacteriophages (“bacteria eaters”)
- Bacteriophage genome can be mapped by following method.
Techniques for the Study of Bacteriophage’s genome
- Viruses reproduce only within host cells; so bacteriophages must be cultured in bacterial cells.
- To do so, phages and bacteria are mixed together and plated on solid medium in a Petri plate.
- A high concentration of bacteria is used so that the colonies grow into one another and produce a continuous layer of bacteria, or “lawn,” on the agar.
- An individual phage infects a single bacterial cell and goes through its lytic cycle. Many new phages are released from the lysed cell and infect additional cells; the cycle is then repeated.
- The bacteria grow on solid medium; so the diffusion of the phages is restricted and only nearby cells are infected.
- After several rounds of phage reproduction, a clear patch of lysed cells (a plaque) appears on the plate (Figure 2.2). Each plaque represents a single phage that multiplied and lysed many cells.
- Plating a known volume of a dilute solution of phages on a bacterial lawn and counting the number of plaques that appear can be used to determine the original concentration of phage in the solution.
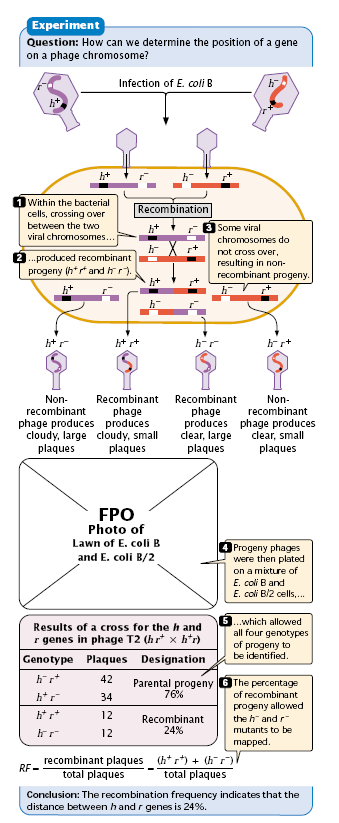
Mapping by Recombination Frequencies –
- The strain B of E. coli can be infected by both h+ and h strains of T2 bacteriophage. In fact, a single bacterial cell can be infected simultaneously by both.
Let us infect a liquid culture of E. coli B with two different mutant T2 viruses
- h r+ and
- h+ r (Figure 2.4)
When this is done in liquid culture, and then plated on a mixed lawn of E. coli B and B/2, four different kinds of plaques appear.
| Genotype | Phenotype | Number of Plaques |
| hr+ | clear, small | 460 |
| h+r | turbid, large | 460 |
| h+r+ | turbid, small | 40 |
| hr | clear, large | 40 |
| Total = | 1000 |
| hm+ | 470 |
| h+m | 470 |
| h+m+ | 30 |
| hm | 30 |
| Total = | 1000 |
- The most abundant (460 each) are those representing the parental types; that is, the phenotypes are those expected from the two infecting strains. However, small numbers (40 each) of two new phenotypes appear.
- These can be explained by genetic recombination having occasionally occurred between the DNA of each parental type within the bacterial cell.
- Just as in higher organisms, one assumes that the frequency of recombinants is proportional to the distance between the gene loci.
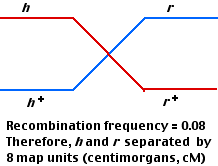
- In this case, 80 out of 1000 plaques were recombinant, so the distance between the h and r loci is assigned a value of 8 map units or centimorgans.
Now repeat coinfecting E. coli B with two other strains of T2:
- hm+ and
- h+m
- Again, 4 kinds of plaques are produced: parental (470 each) and recombinant (30 each).
- The smaller number of recombinants indicates that these two gene loci (h and m) are closer together (6 cM) than h and r (8 cM).
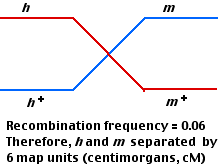
- But the order of the three loci could be either
| mr+ | 440 |
| m+r | 440 |
| m+r+ | 60 |
| Mr | 60 |
| Total = | 1000 |
- m–6–h—8—r or
- h–6–m-2-r
To find out which is the correct order, perform a third mating using
- mr+ and
- m+r
This makes it clear that the
order is m—h—r, not h—m—r. But why only 12cM between the outside loci (m and r) instead of the 14cM produced by adding the map distances found in the first two matings?
Mapping by A Three-Point Cross –
The answer comes from performing a mating between T2 viruses differing at all three loci:
- hmr and
- h+m+r+
(Note: this time one parent has all mutant; the other all wild-type alleles — don’t be confused!)
| Group 1 | hmr | 435 |
| Group 2 | h+m+r+ | 435 |
| Group 3 | h+mr+ | 25 |
| Group 4 | hm+r | 25 |
| Group 5 | hmr+ | 35 |
| Group 6 | h+m+r | 35 |
| Group 7 | hm+r+ | 5 |
| Group 8 | h+mr | 5 |
| Total = | 1000 |
The result: 8 different types of plaques are formed.
- parentals; that is, nonrecombinants in Groups 1 and 2;
- recombinants – all the others
Analyzing these data shows how the two-point cross between m and r understated the true distance between them.
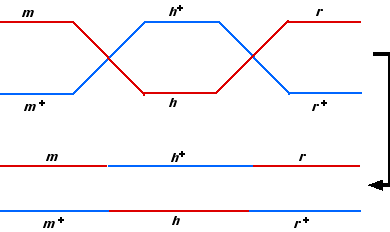
Let’s first look at single pairs of recombinants as we did before (thus ignoring the third locus).
- If we look at all the recombinants between h and r but ignore m (as in the first experiment), we find that they are contained in Groups 5, 6, 7, and 8 -7 giving the total of 80 that we found originally.
- If we look at recombinants between h and m but ignore r (as in the second experiment), we find that they are contained in Groups 3, 4,7, and 8 – giving the same total of 60 that we found before.
- But if we focus only on m and r (as we did in the third experiment), we find that the recombinants are contained in Groups 3, 4, 5, and 6 – giving the same total of 120 as before while the non-recombinants are not only in Groups 1 and 2 but also in Groups 7 and 8. The reason: a double-crossover occurred in these cases, restoring the parental configuration of the m and r alleles.
- Because these double crossovers were hidden in the third experiment, the map distance (12 cM) was understated. To get the true map distance, we add their number to each of the other recombinant groups (Groups 3,4,5, and 6) so 25 + 5 +25 +5 +35 + 5 + 35 + 5 = 140, and the true map distance between m and r is the 14 cM that we found by adding the map distances between h and r (8 cM) and h and m (6 cM).
The three-point cross is also useful because it gives the gene order simply by inspection:
- Find the rarest genotypes (here Groups 7 and 8), and
- the gene NOT in the parental configuration (here h) is always the middle one.



